Peripheral Biomarkers for First-Episode Psychosis—Opportunities from the Neuroinflammatory Hypothesis of Schizophrenia
Article information
Abstract
Objective
Schizophrenia is a disabling disorder of unknown aetiology, lacking definite diagnostic method and cure. A reliable biological marker of schizophrenia is highly demanded, for which traceable immune mediators in blood could be promising candidates. We aimed to gather the best findings of neuroinflammatory markers for first-episode psychosis (FEP).
Methods
We performed an extensive narrative review of online literature on inflammation-related markers found in human FEP patients only.
Results
Changes to cytokine levels have been increasingly reported in schizophrenia. The peripheral levels of IL-1 (or its receptor antagonist), soluble IL-2 receptor, IL-4, IL-6, IL-8, and TNF-α have been frequently reported as increased in FEP, in a suggestive continuum from high-risk stages for psychosis. Microglia and astrocytes establish the link between this immune signalling and the synthesis of noxious tryptophan catabolism products, that cause structural damage and directly hamper normal neurotransmission. Amongst these, only 3-hydroxykynurenine has been consistently described in the blood of FEP patients.
Conclusion
Peripheral molecules stemming from brain inflammation might provide insightful biomarkers of schizophrenia, as early as FEP or even prodromal phases, although more time- and clinically-adjusted studies are essential for their validation.
INTRODUCTION
Schizophrenia is a severe disorder that impairs the person’s entire behaviour and psychosocial functioning. It affects almost 1% of the population worldwide [1], and has a serious socioeconomic impact [2]. Thus far, there is no definite biological explanation for its intriguing symptoms nor its fateful progression. This lack of knowledge in the pathogenesis of schizophrenia makes its diagnosis rely solely on psychopathological descriptions, creating uncertainties and delaying treatment. Furthermore, the compelling need for anticipatory diagnostic strategies was emphasized by studies on high-risk states for schizophrenia that support a significant prognostic impact of early interventions [3-5].
Several techniques have been explored in the search for biological signatures of schizophrenia, comprising genetic, proteomic and metabolomics [6-13], histopathological [14,15], imagiological [16,17], and neurophysiological studies [6,18]. Unfortunately, they could not yet provide a reliable and feasible biomarker for usage in current clinical practice, nor could they support a definite pathophysiological explanation for the disorder and its clinical manifestations.
Interestingly, a growing body of evidence has unveiled a lifelong, insidious immune dysfunction in patients with schizophrenia [19-24] and brought the possibility of tracing its mediators in blood. Therefore, cost-effective diagnostic markers of early-onset schizophrenia, that may even help predict treatment response, are on the horizon [25,26]. However, the increasing literature on inflammation in schizophrenia presents multiple designations and often lays on heterogeneous patient populations, regardless of the disease clinical stage, making it difficult to draw clear-cut conclusions. We, therefore, critically reviewed relevant discoveries on blood markers related to inflammation specifically found in patients at first-episode psychosis (FEP), aiming at seizing promising biomarker candidates for early schizophrenia.
METHODS
We performed an extensive narrative review of literature published in English-language until May 2018. We retrieved references from Pubmed/MEDLINE combining the terms “first-episode”, “schizophrenia”, “inflammat*” and “(bio) marker.” Other relevant articles and printed books were added from cross-referencing. Studies addressing other biomarkers, patients not at FEP or animal models were excluded.
RESULTS
Among 804 references, 18 complied with our scope, reporting three main kinds of markers stemming from inflammation in FEP patients: cytokines, microglial activity markers and tryptophan catabolism products. These with be detailed below and schematized in Table 1.
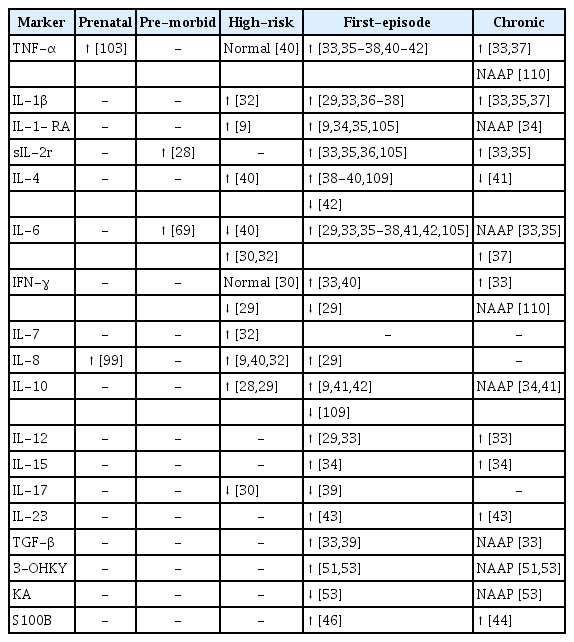
Temporal perspective of peripheral immune biomarkers in Schizophrenia
Biomarkers from cytokines
Only a few biomarker studies adopted prospective assessments and selected drug-naïve acutely ill patients. Before acute illness, asymptomatic first-degree relatives of schizophrenia patients and people experiencing brief or attenuated symptoms (classically described as a prodromal stage) are considered to be at clinical high risk (CHR) for the disease [27]. In relatives at risk, higher levels of serum soluble IL-2 receptor (sIL‐2r) [28] and IL-10 [29] with lower IFN-γ [29] have been reported. In prodromal individuals, increased blood levels of the IL-6 [30], IL-8 [9] and IL-1-receptor antagonist (IL-1-RA) [9], with decreased IL-17 [30], support the hypothesis of a pre-clinically deregulated immune state. The cerebrospinal fluid (CSF) of CHR subjects showed decreased receptors for IL-2 and TNF and elevated IL-8 as well [31]. Also a recent review suggests IL-1β, IL-7, IL-8 and matrix metalloproteinase-8 are predictors of conversion to acute psychosis, with IL-6 failing to prove statistical significance [32].
For FEP, significant increases in serum IL‐1β, sIL‐2r, IL-6, IL-12, TNF-α, TGF‐β, and IFN-γ have been reported by Miller et al. [33], together with reduced IL-10 in acute relapse. In this meta‐analysis, the authors suggested that IL‐1β, IL‐6, and TGF‐β could be “state” markers, given their raised levels in acute episodes followed by normalization under antipsychotic treatment, while IL‐12, IFN-γ, TNF-α, and sIL‐2R were proposed to be “trait markers”, as their levels were maintained after treatment. Recently, other authors reported increased serum levels of IL-1-RA [9,34,35], IL‐1β [29,36,37], sIL‐2r [35,36,38], IL-4 [38-40], IL-6 [29,35-38,41,42], IL-8 [38], IL-10 [9,34,41,42], IL-12 [29], IL-13 [9], IL-15 [34], IFN-γ [40], and TNF-α [35-38,40-42], and decreased levels of IL-17 [39] in drug-naïve FEP patients. Increased serum levels of IL-23, a key modulator of the inflammatory response, were also found in FEP patients and acute relapses [43].
Biomarkers from microglia
Some studies consistently found increased plasmatic concentrations of S100B, a protein marker of glial activity, in patients with schizophrenia [44,45], including in early-stage unmedicated patients [46]. Osteopontin has also been suggested as a marker of microglial activation in blood under experimental exposure to hypoxia and lipopolysaccharide, but to the best of our knowledge, it has not been related to psychosis [47].
Biomarkers form tryptophan catabolism (TRYCAT) products
Tryptophan obtained from our diet is the precursor for serotonin synthesis, hence being largely researched in neuroscience. Some of its metabolites have been found in altered levels in patients with schizophrenia, raising interest about their role [48,49]. Both plasma tryptophan and kynurenine showed decreased in FEP patients [50]. The serum levels of 3-hydroxykynurenine (3-OHKY) showed increased, as well as positively correlated to psychopathology and treatment response [51]. A product of a competing branch in the TRYCAT cascade, Kynurenic acid (KA), did show an increase in the CSF of FEP patients, but not in plasma [48,52,53].
DISCUSSION
The findings in this review bring consistency to the neuroinflammatory hypothesis of schizophrenia, while relying on an explicable theoretical model. We present a schema of this model in Figure 1 to help following our discussion. Extensive data on the potential contribution of inflammation for schizophrenia has been published since Smith’s first immune model [54]. The link between infection and psychosis has been suspected since the 1920s [55], and has found support in epidemiological studies relating schizophrenia either to prenatal [56-59], childhood or adulthood infections [60], irrespective of the infectious agent. Other early environmental determinants, such as low maternal vitamin D, iron or zinc levels, obstetric complications and exposure to certain aromatics, have been implicated in the dysregulation of the immune response [22], following a vulnerability-stress model [61]. These insults could be critical for priming the immune system, making it respond excessively in face of later stimuli, postulated by Keshavan as “second-hits” [62]. The neurodevelopmental hypothesis for schizophrenia [63] hence came to challenge earlier perspectives of neurodegeneration and dementia praecox, postulating it is a multifactorial disease beginning in early life, triggered by successive environmental stimuli to the developing brain of vulnerable subjects. A similar hypothesis is under research for autism spectrum disorders, which share some clinical and immunological features with schizophrenia despite emerging at an earlier age [64].
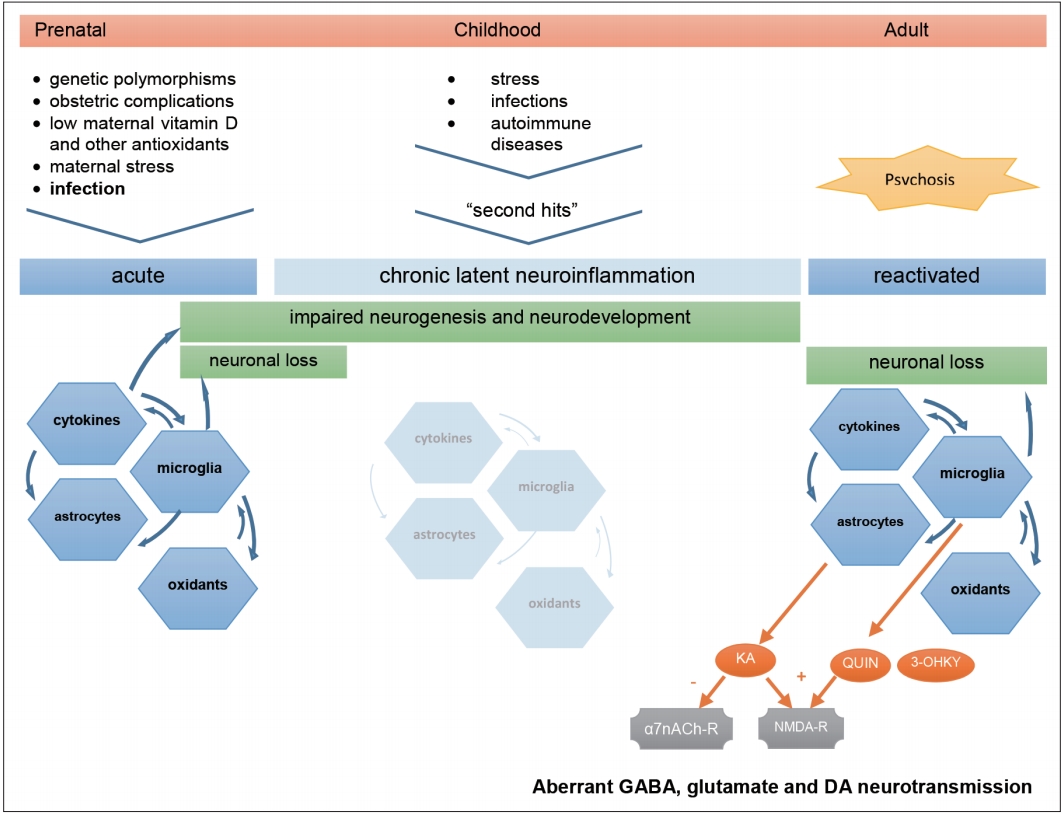
Hypothetical aetiology and pathogenesis of Schizophrenia. Prenatal infection in context of other environmental factors can prime the immune system of already genetically vulnerable offspring. This first insult starts a neuroinflammatory cascade that compromises normal neurodevelopment and is maintained subclinically throughout life, waiting for the next triggers to restart an inflammatory process that, in turn, can lead to neuron damage, aberrant neurotransmission, and clinical symptoms. KA: kynurenic acid, QUIN: quinolinic acid, 3-OHKY: 3-hydroxykynurenine, α7nACh-r: alpha7-nicotinic-acetylcholine receptor, NMDA-r: N-methyl-D-aspartate receptor, DA: dopamine.
Critical immune dysregulation affecting the central nervous system (CNS) involves both T-helper-1 lymphocyte (Th1) and T-helper-2 lymphocyte (Th2) pathways and autoimmune mechanisms [23,24,65-69]. The finding of anti-NMDA-receptor IgG-antibodies in encephalitis with psychotic symptoms emphasized this hypothesis [70], although only IgA and IgM autoantibodies in a minority of patients with schizophrenia has been found so far [71,72]. Genetic research has pointed to various susceptibility genes and defective regulatory microRNA in schizophrenia which are related to immune response [73-76] and family history of the disease was shown to convey a higher risk after prenatal infection [77]. Several contributors to neuroinflammation fit the concept of Keshavan’s second-hits. A defective antioxidant defence system, that prevents neural damage and inflammation in the brain, was found in schizophrenia patients [78,79]. Stress, either in earlylife [80], in the proximity of full-blown psychosis [81] or in later chronic phases [82] has been reported to contribute to the disease through the effects of cortisol on glial activation and cytokine release [83]. Cannabis consumption which has also been widely reported to increase the risk of psychosis [84,85], might also act on cortisol-mediated stress pathways [86]. Finally, the immune involvement in schizophrenia has led to therapeutic trials with anti-inflammatory drugs [87-89] with some promising results already. It should be reminded, however, that neuroinflammation likely underlies other neurological and psychiatric conditions, in which cytokine imbalance and microglial activation are reported, and may be subject to some degree of unspecificity due to confounding factors like obesity, glucose intolerance and tobacco use [35,90-94].
Concerning the biomarker value of cytokines, we should remind they play pleiotropic roles, from immune signalling to regulating early neurogenesis, maturation and neuroplasticity [95]. Increased TNF-α has been implicated in reduced hippocampal neurogenesis [96], as increased IL-6 [97,98] and IL-8 [99] have in reductions of brain volume in schizophrenia. Increased IL-6 levels in children [100] and premorbid risk individuals [101] were later associated to schizophrenia in adult life. Since maternal cytokines can invade fetal CNS [102], we can understand how an immune challenge affecting the mother’s cytokine balance could tamper fetal neurodevelopment [67]. Accordingly, elevated maternal TNF-α [103] and IL-8 [99,104] levels were associated to schizophrenia in the offspring. In this context, inflammation could account for the premorbid impairments in fine motor coordination, memory and social behaviours, which are evident around adolescence in schizophrenia patients [22]. Numerous studies, with cross-sectional design and including patients irrespective of chronicity stage or type of treatment, have reported cytokine alterations in schizophrenia [35,105-107] while others report none [108]. Our findings of elevated IL-1-RA, sIL-2r, IL-6, IL-8, and IL-10, along with lower IFN-γ and IL-17 levels in CHR individuals, support the hypothesis of a pre-clinically deregulated immune state, transitioning to that found in medication-naïve FEP patients, who consistently show increased levels of IL-1-RA, IL‐1β, sIL‐2r, IL-6, IL-8, IL-12, IL-13, IL-15, TNF-α, TGF‐β, and IFN-γ and decreased levels of IL-17. A few discordant reports of lowered IL-4 [42], IL-10 [109], and IFN-γ [29] levels in FEP could reflect some timing differences in sample selection, since an immunological challenge is apparent between T-helper-1 type, proinflammatory cytokines (TNF, IFN-γ, and IL-2) and Thelper-2 type, anti-inflammatory cytokines (IL-4 and IL-10), starting from the early high-risk phase.
Cytokine level normalisation after antipsychotic treatment is in accordance with the hypothesis of their immune modulating action [110], but particular cytokine changes could depend on the drugs used [23], which can be misleading when differentiating chronic “trait” from acute “state markers.” Besides, Song et al. [37], observed normalised cytokine levels rising again after the first weeks of antipsychotic treatment. We therefore defend that such distinction of acute psychosis markers should be based on clinically evaluated symptomatic remission. Correlations with clinical status were found for IL-6 in the metaanalysis by Miller et al. [33], for IL-10 in the meta-analyses by de Witte et al. [34], and for no biomarker at all in the study by Noto et al. [41] Specifically, negative symptoms of schizophrenia (like flattened affect and low volition) were correlated with increased serum IL-4 and decreased IL-10 levels [109], while both positive (delusions and hallucinations) and negative symptoms were correlated to increased IL-1, IL-6, and TNF-α [111].
The role of cytokines in schizophrenia becomes even more relevant if we consider their effects on microglia, the brainresident macrophages. Microglia become activated by proinflammatory cytokines in response to immune challenges [82], releasing cell-damaging reactive oxygen and nitrogen species and other cytokines. They also enhance TRYCAT towards detrimental end-products [23]. Early-life microglial activation, triggered by infection or other insults, could initiate priming of the immune system, maintaining low-level inflammation and making it prone to exaggerated response to later challenges, thus increasing the risk for schizophrenia [83,112]. In accordance, microglial activity has been found increased in the brains of patients with schizophrenia in imagiological studies, namely in the anterior cingulate cortex, mediodorsal thalamus [113] and hippocampus [16]. Microglial overactivation can be associated with excessive synaptic pruning and at least sub-lethal apoptotic activity [114], relating to regional brain atrophy in first-episode psychosis and acute relapses [115]. Not surprisingly, elevated plasmatic S100B levels are found in FEP patients, making the protein suitable for psychosis biomarker. Further plasmatic molecules reflecting microglial activity should be sought to increase diagnostic accuracy.
Finally, tryptophan catabolism byproducts mediate the aforementioned immune changes in schizophrenia, through direct interference with neurotransmission. IL-1β, IL-6, TNF-α, IFN-γ, and IL‐18 induce microglial enzyme indoleamine-2, 3-dioxygenase (IDO), whereas IL-1β and stress-driven cortisol induce astrocytic enzyme tryptophan-2,3-deoxygenase (TDO) [116]. TRYCAT cascade under IDO releases excitotoxic NMDA-receptor agonists quinolinic acid (QUIN) [117] and 3-OHKY, while under TDO it releases KA. KA is an antagonist to NMDA and α7nACh receptors, impairing inhibitory activity of subcortical GABA-ergic interneurons [118]. This ultimately leads to unbalanced mesolimbic dopamine neurotransmission that can elicit psychotic symptoms [49,119]. Likewise, while the dopaminergic hypothesis for schizophrenia has prevailed for decades, attention to the hyperglutamatergic activity in the frontal cortex, secondary to decreased subcortical inhibition, has raised [120]. This perspective is more in accordance with the hypothesis of functional dysconnectivity in schizophrenia [121]. The value of KA as a feasible biomarker for psychosis is still questionable, however. Its levels were found elevated in their CSF but not in peripheral blood [48,122]. Besides, considerable part of brain kynurenine is produced systemically and KA cannot normally traverse the blood-brain barrier. On the other hand, TRYCAT cascade showed tendency to lean to the 3-OHKY branch, this metabolite being reported elevated in peripheral blood of FEP patients more consistently. As a matter of fact, excitotoxic and neurotoxic TRYCAT end-products such as 3-OHKY, QUIN and glutamate can cause neuronal loss and explain neuroprogression in schizophrenia [51].
CONCLUSION
In conclusion, this is the first study, of our knowledge, addressing serum biomarker candidates in relation to the whole natural history of the disease. Cytokine level changes are the most widely reported so far in FEP and appear in a continuum from high-risk stages, in accordance with the hypothesis of a progressive inflammatory process, which also allows an anticipated diagnosis. To help surpass their possible limitation of specificity for the disease, other molecules (such as inflammation-related microRNA) should be part of a comprehensive biomarker panel, in order to assist the diagnostic rationale in clinical practice. The present review is subject to selection and publication bias itself. In the future, prospective studies, monitoring these molecules in parallel to clinical presentation, with longer follow-up periods, are warranted for validation. This represents a crucial initial step for early and reliable diagnosis of schizophrenia and prognostic enhancement.
Acknowledgements
This work has been conducted with the support of the Portuguese Foundation for Science and Technology.