INTRODUCTION
Substantial advances in research methods offer new insights into the neurobiological mechanisms of psychiatric disorders. An accurate understanding of the molecular, cellular, and neural pathways of human psychiatric illnesses requires the investigation of human brain tissue. Unlike other human organs, brain tissue cannot be assessed by using biopsy samples of patients with psychiatric illness. Therefore, limitations exist on research concerning the neurobiology of human psychiatric disorders.
Brain samples obtained during an autopsy are critical for investigation of human psychiatric disorders. However, these samples represent disease end points and reveal little about disease initiation and progression, and are also difficult to collect postmortem. Likewise, neuroimaging studies of patients may measure consequences of the disease state rather than its origin. Lack of information about the neural circuit and whole brain networks are also part of the problem. Animal models are challenging to interpret and translate to patients because of species-specific differences between the human and animal brain. Additionally, the lack of translatable animal models, particularly for the disorders in which higher function is involved, and the reliance on behavioral models may also be limiting.1 Furthermore, genetics research requires recruitment of a significant number of patients for more accurate results. Finally, peripheral tissue samples from patients may not be representative of neural tissue that is related to disease development. In order to overcome these limitations, the generation of a new study model is necessary for developing the knowledge of the neurobiology of human psychiatric diseases.
Human induced pluripotent stem cells (hiPSCs) were first generated in 2007 by Yamanaka et al. hiPSCs were successfully derived from human fibroblasts, and this was the first conversion of somatic cells to human embryonic stem cells (hESCs). hiPSCs and hESCs share common characteristics including the capacity for self-renewal, pluripotent differentiation potential, as well as morphology. However, hiPSCs have the advantage of bypassing the ethical issues of destroying embryos to obtain hESCs and immunological hurdles from heterologous cells. Induced pluripotent stem cells (iPSCs) are adult pluripotent stem cells generated from somatic cells by the introduction of a set of transcription factors linked to pluripotency using either the classic virus-based methods or other methods developed to avoid integration of foreign DNA into the host genome.23 Ectopic expression of the four transcription factors Klf-4, Oct4, Sox2, and c-Myc induces reprogramming of somatic human cells to a pluripotent state.24 These hiPSCs have characteristics similar to those of hESCs, derived from the inner cell mass of the embryonic blastocyst, and together hiPSCs and hESCs are referred to as human pluripotent stem cells. Patient-derived hiPSCs can then be differentiated into a wide range of somatic cell types including neuronal cells by adding a specific combination of growth factors under special culture conditions.5
Although ethical issues specific to the use of iPSCs and the potential of iPSCs to accumulate point mutations during culture may exist, the applications of this technology hold tremendous promise for studying and treating brain psychiatric disorders. Importantly, the advent of technology for somatic cell reprogramming now enables in vitro modeling of human brain cells in various psychiatric disease states using patient-derived cells. Because hiPSCs can be derived from adult patients after the development of psychiatric disease, they represent a potentially limitless source of human brain cells with which to study disease, even without knowing which genes are interacting to produce the disease state in an individual patient.6 In addition, reprogramming was initially established using dermal fibroblasts and integrating viral vectors, however, recent advances allow derivation of hiPSCs from cell types accessible by less invasive methods, such as from keratinocytes,7 peripheral blood T lymphocytes,89 and exfoliated renal epithelial cells found in urine samples.1011 In combination with efficient transgene-free reprogramming,12 these improvements facilitate the generation of hiPSCs from patients enrolled in clinical trials or studies. The iPSC approach to studying psychiatric disorders has enormous potential for many applications, including disease-specific cellular models, disease mechanisms, platforms of drug discovery, cell-based therapy, and autologous sources for cell replacement therapy.
iPSCs provide a novel way to study molecular and cellular mechanisms of neuronal differentiation, maturation, and degeneration. Disorders associated with the origin of neurodevelopment and neurodegeneration have been preferentially studied, such as autism spectrum related disorders, schizophrenia, bipolar disorder, and dementia. In this article, we review the current use of iPSCs to study such psychiatric disorders.
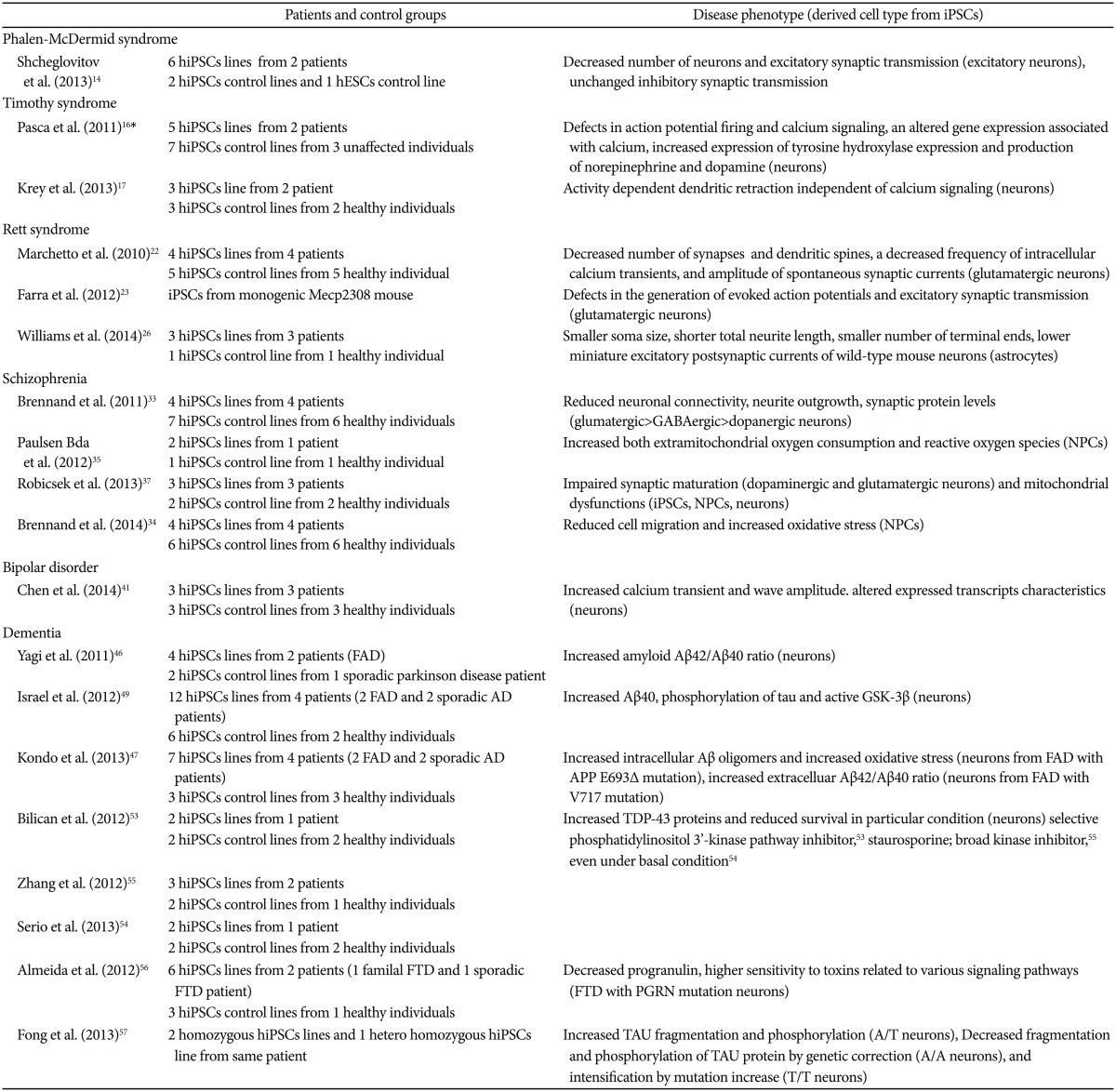
SUMMARY OF PREVIOUS STUDIES ON PSYCHIATRIC DISEASES INVOLVING iPSCs
Phelan-McDermid syndrome
Phelan-McDermid syndrome (PMDS) is a neurodevelopmental disorder, which presents symptoms such as seizures, intellectual disability, impaired speech, and increased risk of autism spectrum disorders (ASDs) by deletion of the SH3 and multiple ankyrin repeat domains 3 (SHANK3) gene on chromosome 22q13.3.13 Generated hiPSC-derived neurons from PMDS patients showed significant deficits in excitatory synaptic transmission due to both an incorrect number of excitatory synapses and reduced expression of glutamate receptors in contrast to unconverted inhibitory synaptic transmission. hiPSC-induced neurons in PMDS show that loss of a copy of SHANK3 downregulated the expression of the SHANK3 protein isoform. Several drugs including trichostain A, valproic acid, nifedipine, and IGF2 were tested to restore the synaptic deficits in PMDS based on previous studies. However, only treatment of neurons differentiated from PMDS-derived hiPSCs with IGF1 showed restored excitatory synaptic transmission demonstrated by an increase in the number of synaptic α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-Methyl-D-aspartate (NMDA) receptors. IGF1 treatment with derived neurons from PMDS patients encourages formation of a class of synapses containing postsynaptic density protein 95 (PSD95) but lacking in SHANK3 associated synapses.14
Timothy syndrome
Timothy syndrome (TS) is a multisystemic disorder caused by a mutation in the CACNA1C gene, which encodes voltage gated calcium channels such as Ca (V) 1.2. Clinical manifestations associated with this disease include syndactyly, congenital heart disease, cardiac arrhythmia, and autism.15 Transformed hiPSCs-derived neurons from two type-1 TS patients presented wider action potential and increased intracellular calcium levels, which indicate defects in action potential firing and calcium signaling. In addition to altering genes related to calcium dependent regulation, TS mutation caused upregulated expression of tyrosine hydroxylase leading to increased production of norepinephrine and dopamine. Decreased neuronal expression in the lower cortical layer, such as the fractional callosal projection, and increased upper neuronal expression, such as subcortical structure, was also demonstrated with TS mutation. Roscovitine, a cyclic-dependent kinase inhibitor and atypical L-type channel blocker, reduced the ratio of tyrosine hydroxylase neurons in patients with TS, which could restore increased expression of tyrosine hydroxylase in the cells of individuals with TS.16 Krey et al.17 found that mutant TS Cav 1.2 channels result in activity-dependent dendrite retraction in both rat and human neurons. However, this phenomenon was not mediated by a calcium dependent mechanism, but rather by insufficient recruited levels of the small GTP-binding protein Gem by the mutant TS Cav 1.2 channel, leading to excessive ectopic activation of RhoA (Rashomolog gene family, member A, a small GTPase) hence dendritic retraction.
Rett syndrome
Rett syndrome (RTT) is one of the most prevalent female neurodevelopment disorders, with an incidence of 1 in 10,000-15,000 female births. Patients with RTT show normal development until 6-18 months after birth and then present symptoms including severe mental retardation, absence of speech, stereotypic hand movements, epileptic seizures, encephalopathy, and respiratory dysfunction.18 Mutations in the X-linked gene encoding methyl CpG binding protein2 (MECP2) were identified as the most common (over 95%) genetic cause of RTT.19 Moreover, cyclin-dependent kinase-like 5 (CDKL5)20 and forkhead box G1 (FOXG1) on chromosome 14 are known to be related to RTT.21
While no differences were observed in neurogenesis, glutamatergic neurons derived from the hiPSCs of four RTT patients had fewer synapses, reduced spine density, smaller soma size, a reduction in the transient rise of intracellular calcium levels of active synapse, as well as a decrease in frequency and amplitude of spontaneous postsynaptic currents relative to controls. Taken together, they suggest a deficiency in neuronal network connectivity. Decreased synapse number in some clones and decreased MECP2 levels in RTT neurons were improved following treatment with IGF1 and low dosage of gentamicin, respectively.22 iPSC-derived neurons from heterozygous Mecp2308 mice showed significant dysfunction in the generation of evoked action potentials, which included decreased action potential amplitude, decreased number of action potentials, and prolonged rise time, decay time, and duration. Diminished peak inward currents and higher input resistance was also observed compared with wild-type iPSCs-derived neurons. Furthermore, reduced frequency of miniature excitatory postsynaptic currents in hemizygous mutant mouse iPSCs induced neurons present deficits in excitatory synaptic transmission.23
Moreover, recent RTT hiPSCs studies provide genetic mechanisms involving the MECP2 regulatory (transcriptional regulation, modulation of the expression of mitochondria related genes, and modulation of synaptic expression of a dysbindin Bloc Network) pathway in undifferentiated cells.2425 In addition, differentiated astrocytes from the hiPSCs from three RTT patients have a negative effect on the morphology and function of neuronal growth in wild-type mice, as also observed in RTT human autopsy samples and in RTT mouse models. Short-term treatment with IGF1 or GPE (a peptide containing the first 3 amino acids of IGF1) increased the wild type neuronal soma size caused by RTT mutant hiPSCs derived astrocytes.26 Clones of CDKL5-mutated iPSCs induced neurons from a female and a male RTT patient demonstrated the possibility of these iPSCs to serve as ideal experimental controls.27 HiPSCs from RTT patients with FOXG1 are yet to be reported.
Schizophrenia
Schizophrenia (SCZ) is a devastating mental disorder2829 characterized by the following three symptom categories: Positive symptoms including hallucinations and delusions, negative symptoms such as apathy and anhedonia, and cognitive dysfunction in attention and working memory. It is widely accepted that the pathophysiology of SCZ involves neurodevelopmental processes, which presumes that abnormal neurodevelopment, including impaired synaptic pruning, precedes the onset of prominent psychotic symptoms.303132 Previous studies, including postmortem, brain imaging, pharmacological, genetic, and animal studies, have provided valuable insight into common phenotype and neuronal pathology of disease despite the heterogeneity in clinical manifestation. However, these studies have revealed less about disease initiation and progression due to implicit limitations.
Reprogrammed fibroblasts from SCZ patients can be differentiated into neurons via hiPSCs. SCZ hiPSCs neurons showed reduced neuronal connectivity in conjunction with decreased neurite number, PSD95-protein levels, and glutamate receptor expression. Gene expression profiles of SCZ hiPSCs neurons identified altered expression of many components of the cAMP and WNT pathways, which are closely related to psychiatric disorders. Key cellular and molecular elements were improved following a 3-week treatment of SCZ hiPSCs neurons with the specific antipsychotic Loxapine. However, this study had limitations related to the heterogeneity of patients, had a small sample size, and limited patient profile.33 A subsequent research study showed that neural progenitor cells (NPCs), which are a minimally differentiated form of neurons, from SCZ hiPSCs have aberrant cell migration and demonstrate increased oxidative stress.34
Paulsen et al.35 reported that only NPCs, not fibroblast cells, and iPSCs derived from SCZ hiPSCs presented a two-fold increase in extra-mitochondrial oxygen consumption as well as an increase in the level of reactive oxygen species. Furthermore, the mood stabilizer, valproic acid reverted the altered extra mitochondrial oxygen consumption and reactive oxygen species of SCZ NPCs to normal levels. Further studies also showed increased levels of zinc and potassium from SCZ hiPSCs derived NPCs, which were improved by valproic acid treatment.36
hiPSCs derived from the hair follicles of SCZ patients demonstrated the presence dopaminergic neurons with the inability to differentiate, glutamatergic neurons with maturation difficulties. Mitochondrial dysfunctions such as impaired mitochondrial respiration, sensitivity to dopamine induced inhibition and dissipation of mitochondrial membrane potential (Δψm) was also observed.37 Yu et al.38 differentiated SCZ hiPSCs into dentate gyrus NPCs that presented lower expression levels of hippocampal neurogenesis. Moreover, SCZ-hiPSCs-derived neurons showed reduced levels of neuronal activity and spontaneous neurotransmitter release.
Bipolar disorder
Bipolar disorder (BD) is an affective disease characterized by alternating episodes of mania and depression. The heritability of BD is estimated at 79-93%, and studies have found candidate genes and genetic mechanisms associated with BD, despite its complex etiology.39 However, BD is typically diagnosed in the late teens to twenties, which presumably presents the influence of abnormalities during the neurodevelopmental period as well as progressive neuropathic changes.40 Although there were no transcriptional differences between BD hiPSCs and control hiPSCs, BD hiPSCs induced neurons expressed increased membrane receptors and ion channels compared with control neurons. In addition, BD hiPSCs induced neurons expressed genes associated with an early ventral CNS fate, while control neurons expressed transcripts characteristic of a dorsal telencephalic fate. Furthermore, BD hiPSCs induced neurons had a higher calcium transient and wave amplitude than controls, which can be restored by lithium pretreatment.41
Alzheimer's disease
Alzheimer's disease (AD) is the most common neurodegenerative disease and the most common cause of dementia with pathologic findings including reduced synaptic connectivity, neuronal loss, and intracellular neurofibrillary tangles composed of the Aβ amyloid fragments of the amyloid precursor protein (APP) and hyper-phosphorylated Tau protein.42 Mutation in APP and Presenilin 1, 2 (PS 1, 2, which are two components of the γ-secretase complex) genes are common causes of Familial Alzheimer's Disease (FAD).4344
By using hiPSCs, human neuronal models of dementia derived from patient tissue can be generated.45 Differentiated hiPSCs neuronal cells from a FAD patient with mutation in the PS1 and PS2 genes showed an increased Aβ42/Aβ40 ratio as seen in post mortem study from FAD patients.46 The longer Aβ peptide form, Aβ42, appears to be more toxic to cells at higher concentrations, therefore an increased Aβ42/Aβ40 ratio may promote toxicity.42 Moreover, derived neurons from hiPSCs of FAD patients with APP mutation47 and induced PS1 mutation from unaffected individuals48 also presented this increased extracellular Aβ42/Aβ40 ratio phenomenon. Generated hiPSCs derived cortical neurons from three AD patients, including one sporadic AD patient, displayed increased Aβ40 expression, phosphorylation of the tau protein, and activated glycogen synthase kinase 3β (aGSK-3β) compared to controls. However, there were no significant differences in synapse number, neuronal survival, and electrophysiological activity even after several weeks.49
Derived hiPSCs neurons from a FAD patient with APP mutation showed accumulated intracellular Aβ oligomers in contrast to very low extracellular levels of the Aβ form, leading to endoplasmic reticulum (ER) stress and oxidative stress, which might contribute to AD pathogenesis. This suggests the idea of a two type of AD classification: the extracellular Aβ type and the intracellular Aβ type. Transformed neurons from one of the sporadic AD patients also showed increased intracellular Aβ oligomers, which could be rescued by β-secretase activity inhibitors.47 These findings are in line with the notion of genomic significance in sporadic AD as well as the effect of environmental factors and aging.
Despite such improvements, it is still difficult to make a sporadic AD model, which is the most common cause of AD, due to a combination of multiple genetic factors (Apolipoprotein E isoform E4, CR1, Clusterin and SORL1) and non-genetic factors such as environment stressors.50
Fronto-temporal dementia
Fronto-temporal dementia (FTD) is the second most common presenile dementia after AD, and is characterized by neuronal and synaptic loss in the frontal cortex. Clinically it is associated with more prominent deficits in higher cognitive function compared with AD.51 Mutations in the transactive response DNA binding protein-43 (TDP-43) locus, progranulin (PGRN) gene, microtubule associated protein tau (MAPT) gene, intronic hexacucleotide repeats, and expansions within the C90RF72 locus can all lead to familial cases of the disease.52 Mutant TDP-43 hiPSCs derived neurons have higher levels of the TDP-43 cytoplasmic protein as well as reduced survival.535455 Mutant neurons displayed reduced survival in the presence of a selective phosphatidylinositol 3'-kinase pathway inhibitor53 and staurosporine, a broad kinase inhibitor,55 even under basal conditions.54
Neurons derived from an FTD patient with a PGRN mutation were more vulnerable towards toxins, such as tunicamycin (protein N-glycosylation inhibitor) and lactamycin (proteasome inhibitor). Furthermore, these derived neurons revealed increased sensitivity to straurosporine, two PI3K/Akt inhibitors, and a MEK/MAPK inhibitor, which implicates the underlying pathogenesis of human PGRN mutation. These results, except endoplasmic reticulum stress induced by tunicamycin, were rescued by ectopic PGRN expression. Interestingly, the FTD-PGRN-hiPSCs-neuron cells had sustained TDP-43 mislocalization.56
Fong et al.57 generated replaced hiPSCs-derived neurons (A/A) from a patient with a heterozygous MAPT (A/T) mutation using zinc-finger technology for mutation correction. After mutation correction, recovered neurons (A/A) had decreased TAU fragmentation and phosphorylation, whereas neurons intensified with homozygous MAPT mutation (T/T) had increased phosphorylation and enhanced TAU fragmentation.
Several hiPSCs studies have focused on the hexanucleotide CRORF72 GGGGCC repeat expansion upstream of the coding region of C9ORF72 and suggested accumulation and disturbed RNA metabolism as a contributing pathogenic factor in C9ORF72 FTD.585960 The results of the research above are summarized at Table 1.
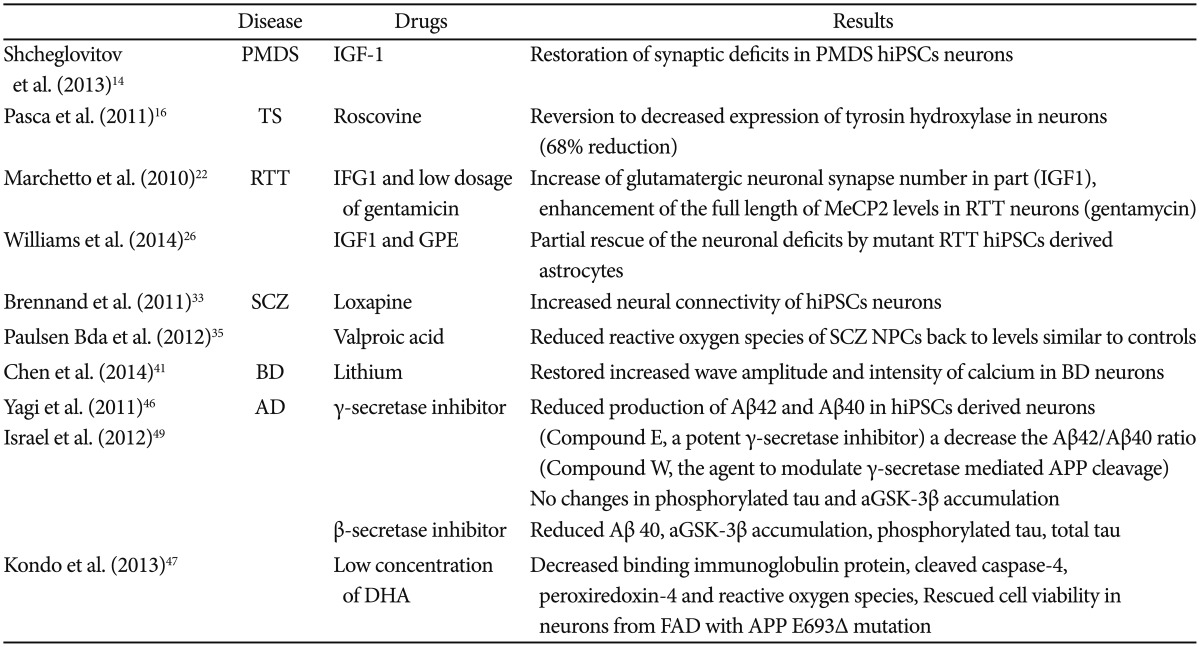
Drug studies involving hiPSCs
The lack of representative animal models, reliable biomarkers, and precise mechanisms of psychiatric diseases in addition to technical challenges in obtaining proper samples from patients have all presented significant hurdles to psychiatric drug discovery. The advent of new technology, such as hiPSCs that reflect the genetic signature of the patients from which they are derived from, could overcome these obstacles. Many advanced drug studies using hiPSCs from diverse psychiatric patients have been conducted (Table 2) and it appears that these studies have two general mainstreams so far.
First, recovery of a patient's cellular phenotypes by applying existing drugs approved in a clinical setting or putative agents related to molecular mechanisms of disease pathogenesis, which then provides evidence that supports the validity of a cellular model for a psychiatric disorder. For instance, although both β and γ-secretases reduced the level of Aβ40, only β-secretase inhibitors affected phosphorylated TAU and GSK-3β in hiPSCs derived neurons from AD patients. This may suggest underlying mechanisms of APP processing by β-secretase and γ-secretase activity and genetic factors associated with sporadic AD.49
Second, amelioration of the altered phenotypes of patient's hiPSCs using new classes of drugs that demonstrate therapeutic efficacy towards known or novel targets enable researchers to establish valuable drug screening and drug discovery tools. Most of the studies at present actually belong to the former category, since the latter faces limitations,61 which could be overcome in the near future by technological advances (mentioned later).
DISCUSSION
Limitations of previous studies on iPSCs
A number of limitations in former iPSCs studies exist. First, several previous studies stand on the basis of the cell banks, which provide only basic diagnostic and demographic information, leading to limited patient profiles. Second, due to their small sample size (typically 1-4 patients), the results of previous studies cannot be assumed as representative of the larger patient population. Third, some studies were conducted with epidemiological differences between experimental patients and controls and heterogeneity of patient and control cohorts, even without controls. Fourth, there are still no effective experimental methods to control inter-patient and intra-patient variability, including experimental variability, scalability of hiPSCs generation and neural differentiation; although many groups are pursuing strategies to overcome these difficulties. In addition to previous studies, there are still unexplored fields in psychiatric disease to which hiPSCs technology can be applied, such as major depression and attention deficit hyperactivity disorder with genetic risk factors.
Future directions
There could be latent practical harm and obstacles involved in obtaining more biopsy samples from fibroblast or adipose tissue cells of patients who suffer from anxiety and worry. Generating hiPSCs from hair follicles, urine, and blood may be safer and convenient options for recruiting more patients for technologically advanced studies. Unlike well-studied monogenic neurodevelopmental disorders such as Rett syndrome, most psychiatric diseases are attributed to multiple levels of etiologic causes, which could be a potential obstacle to successful study. Furthermore, previous studies involving psychiatric patients with early onset history and significant hereditary family history was successful to some extent;33 however, rare inclusion criteria make it challenging to represent a larger patient population and be applied to sporadic psychiatric disease.
Therefore, using a patient cohort with a shared clinical phenotype or comparing patients with or without phenotypes for selecting subgroups such as pharmacological responses can help overcome inter-patient variability.62 For instance, one study used hiPSCs from schizophrenia patients with DISC gene to make a homogeneous patient population design63 whereas another study included hiPSCs from one schizophrenia patient, which was identified as clozapine resistant.35 Furthermore, investigators who wish to collect a larger sample size should consider more refined clinical phenotypic characterizations combined with various information such as previous medical history, family history, drug history, genetic history, neuroimaging history, and even neuropsychological assessment of patients, if possible. Using these numerous patient information, researchers might be able to present a more integrated approach to hiPSCs disease modeling.
The generation of hiPSCs is now becoming a more common study design tool in psychiatry. However, differentiation of these hiPSCs into neuronal types of interest remains challenging though it is important, as correct development is essential to the neocortex, especially with unaffected individuals. Therefore more standardized protocols of differentiation should be generated. Because iPSC-based technology has yet to overcome its intrinsic constraints as mentioned above, one alternative to reprogramming strategies for differentiation of neurons is skipping cellular reprogramming and differentiation, which require at least a few months for the conversion of fibroblasts, and going directly to neurons,6465 or using exogenous neuronal transgenes.66 However, it is important to note that direct conversion skips the normal developmental process, which may be a critical time of disease pathogenesis.67
Future approaches for drug studies will explore not only finding drugs that confirm known disease models or drug effects but also finding unrecognized pathophysiological mechanisms of psychiatric disease, which could lead to drugs targets using more efficient methods such as shorter time, larger populations of homogeneous cell types, and lower financial costs. To achieve this, a more standardized system such as high-throughput screening and sophisticated cell purifying methods using specific cell surface markers should be evolved. Recent advances in genetic manipulations provided some opportunities to produce homogeneous and stable populations of late cortical progenitors from hiPSCs, which could be amplified and differentiated preferentially into glutamatergic neurons. Moreover, this method shortens the timeframe for generating neurons from hiPSCs and enables investigators to make larger banks that can be stored and frozen.68
CONCLUSION
hiPCSs reprogrammed from patient's somatic cells present a novel tool to study psychiatric disorders which conventionally lack methodological approaches such as translatable animal models, reliable biomarkers of therapeutic efficacy69 and ethical strategies for directly obtaining, thus spreading the range of treatment option. Since iPSCs retain the genetic composition of the patient and presumably reproduces the alteration of those of patients compared to controls, information regarding changes of synaptic development and function such as synaptic transmission, as well as alteration in intracellular signaling and neuronal maturation have been found in most studies of hiPSCs derived neurons so far. It is expected that hiPSCs will serve as a useful tool for studying the neurodevelopmental process of psychiatric disease, and will also be useful in both drug screening and drug development.