A Novel Bio-Psychosocial-Behavioral Treatment Model of Panic Disorder
Article information
Abstract
To conceptualize a novel bio-psychosocial-behavioral treatment model of panic disorder (PD), it is necessary to completely integrate behavioral, psychophysiological, neurobiological, and genetic data. Molecular genetic research on PD is specifically focused on neurotransmitters, including serotonin, neuropeptides, glucocorticoids, and neurotrophins. Although pharmacological interventions for PD are currently available, the need for more effective, faster-acting, and more tolerable pharmacological interventions is unmet. Thus, glutamatergic receptor modulators, orexin receptor antagonists, corticotrophin-releasing factor 1 receptor antagonists, and other novel mechanism-based anti-panic therapeutics have been proposed. Research on the neural correlates of PD is focused on the dysfunctional “cross-talk” between emotional drive (limbic structure) and cognitive inhibition (prefrontal cortex) and the fear circuit, which includes the amygdala-hippocampus-prefrontal axis. The neural perspective regarding PD supports the idea that cognitive-behavioral therapy normalizes alterations in top-down cognitive processing, including increased threat expectancy and attention to threat. Consistent with the concept of “personalized medicine,” it is speculated that Research Domain Criteria can enlighten further treatments targeting dysfunctions underlying PD more precisely and provide us with better definitions of moderators used to identify subgroups according to different responses to treatment. Structuring of the “negative valence systems” domain, which includes fear/anxiety, is required to define PD. Therefore, targeting glutamate- and orexin-related molecular mechanisms associated with the fear circuit, which includes the amygdala-hippocampus-prefrontal cortex axis, is required to define a novel bio-psychosocial-behavioral treatment model of PD.
INTRODUCTION
Panic disorder (PD) has a lifetime prevalence ranging from 1 to 3% in the general population and a prevalence ranging from 3.0 to 8.3% in clinical settings [1-4]. The Global Burden of Diseases, Injuries, and Risk Factors study indicated that anxiety disorders including PD have contributed to 1% of all disability-adjusted life years and 3.5% of all of years lived with disability worldwide. In addition, the disease burden caused by anxiety disorders is only second to that due to depressive disorders among all mental disorders [5,6]. Even with optimal treatment consistent with current clinical guidelines, half or more of patients with PD have had suprathreshold or subthreshold panic symptoms in the unremitted state [7].
The Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5) was revised in order to accommodate a paradigm shift from using a categorical approach to using a dimensional approach in psychiatric nosology and taxonomy. However, the categorical concept of PD still remains in the DSM-5. Highlights of the changes to diagnostic criteria for PD from the DSM-IV to DSM-5 are as follows: 1) distinction of agoraphobia from PD and 2) defining panic attacks as a specifier. The diagnostic criteria for PD have been revised based on the considerable prevalence of agoraphobia without PD in the community setting, significant functional declines in patients with agoraphobia without PD, significant differences between agoraphobia and PD in terms of sociodemographic and clinical characteristics, and the observation that panic attacks are not limited to patients with PD. Neurobiological perspectives regarding cognitive models can be more emphasized in descriptions of PD, with an emphasis on “the amygdala and related structures” in “current neural systems models” and a potential “association between PD and the catechol-O-methyltransferase gene in females.” In the DSM-5, a diagnosis of PD is operationally defined as the fulfillment of both recurrent unexpected panic attacks (Criterion A) and the existence of one or more of the following persistent panic attack-related conditions for at least one month: concern, worry, and behavioral change (Criterion B) [8-13].
PD was initially modeled as being neither exclusively biologically determined nor psychologically determined. This PD can be treated effectively using both pharmacotherapy and cognitive-behavioral therapy (CBT) [14]. Thus, according to the original hypothesis by Gorman et al. [15], PD was modeled as having three main panic symptoms: panic attacks and stress response, anticipatory anxiety and heightened fear, and phobic avoidance and emotional regulation. These symptoms were distinctively associated with neural substrates including loci in the brainstem and hypothalamus, the limbic system, and the prefrontal cortex, respectively. Moreover, tricyclic antidepressants (TCAs) are considered distinctively effective in treating panic attacks at the brainstem level, benzodiazepines or relaxation and ventilation techniques are distinctively effective in treating anticipatory anxiety at the limbic level, and CBT is distinctively effective in treating phobic avoidance (agoraphobia) at the prefrontal cortex level. According to the revised hypothesis by Gorman et al. [16], the essential neural substrate of PD is the dysfunctional “cross-talk” between emotional drive (limbic structures) and cognitive inhibition (prefrontal cortex) [17,18]. In addition, the biopsychosocial model of PD, which is currently criticized for its eclectic framework, has ambiguous characteristics used for the determination of scientific and therapeutic priorities among the biological, psychological, and social factors associated with psychiatric disorders [19-22]. Here we aimed to review and discuss a novel bio-psychosocial-behavioral treatment model of PD with a multilevel framework from the perspective of integrationism. First, the specific molecular genetic basis for PD is reviewed and investigated. Second, the current status and limitations of psychopharmacological interventions for PD are reviewed and novel investigational therapeutics for PD are discussed. Third, the neuroanatomical correlates of PD and CBT for PD are reviewed and discussed.
MOLECULAR GENETIC BASIS FOR PANIC DISORDER
There are more than 20 identified candidate genes for susceptibility to PD affecting different neurotransmitter systems including the serotonin, norepinephrine, adenosine, γ-aminobutyric acid (GABA), and glutamate systems, different neuropeptides, and the hypothalamic-pituitary-adrenal axis. However, findings related to the identified genes for PD have not been replicated in larger cohorts and in different populations, as the samples used in those studies have rendered them underpowered for the detection of risk variants associated with moderate effect sizes for more than 500 cases and more than healthy controls and marker coverage has not been sufficient to capture the locus-specific pattern of linkage disequilibrium in most candidate gene studies of PD. Studies using mouse models with genetic manipulations of PD candidate genes are required to further elucidate the roles of those genes in regulating behavior [17,23,24].
Serotonin
An animal study indicated that inactivation of the Lmx1b gene in the raphe nuclei in 5-hydroxytryptamine (5-HT)-deficient mice led to near complete 5-HT depletion in the brain and enhanced contextual fear memory [25]. Because of the potential role of central 5-HT in protecting the hippocampus from stressful events and reducing the recollection of fear memory, a decreased hippocampal volume may be associated with susceptibility to PD [26]. Thus, hippocampal dysfunction in associative learning and context encoding may be related to phobic avoidance, learning discrimination, and contextual fear memory in PD. In addition, mice with knockout of the Htr1a serotonin receptor display enhanced contextual fear memory [27]. Because of the inhibitory nature of the 5-HT1 receptor family, depletion of this receptor family may increase serotonin release in areas of serotonin production. This may in turn affect the availability of serotonin in target regions [28]. In line with the above speculation, a marked reduction in 5-HT1A receptor binding in the anterior and posterior cingulate cortices was reported in a positron emission tomography (PET) study of patients with PD [29].
Mice with knockout of the serotonin transporter (5-Htt) display impaired contextual fear conditioning. This represents deficient serotonin clearance in the synapse and excess serotonin in the synaptic cleft [30]. When using the less hippocampus-dependent cue fear conditioning paradigm, conditioned fear extinction has been considered to reflect the formation of a new safety memory in the context of extinction learning [31]. Significant alterations in the ventromedial prefrontal cortex and hippocampus in response to the extinguished and non-extinguished stimuli during extinction recall have been positively correlated with the magnitude of extinction memory in humans [32]. A 5-Htt knockout mice study reported alterations in dendritic morphology in infralimbic pyramidal neurons and an increase in spine density in the basolateral amygdala [31]. The above findings can be explained as follows: An increased excitatory drive resulting in amygdala hyperactivity may be involved in the increase in spine density in the basolateral amygdala, and an adaptive response to decrease the hyperactivity may be involved in the increased dendritic extent of the infralimbic neurons [33]. However, impaired fear memories have not been reported in rodent models with alterations in the genes for the serotonin receptors Htr2a and Tph2 [34-36]. In conclusion, the processing of hippocampus-dependent fear memories has been shown to be affected by manipulations that result in alterations in serotonergic activity.
Neuropeptides
Among the dozens of neuropeptides, neuropeptide Y (NPY) has been of interest for its role in anxiety-related disorders. This is because NPY is specifically found in the hippocampus, amygdala, and cortex, and NPY receptors (Y1, Y2, Y4, and Y5) are particularly expressed in the hippocampus [37,38]. Considering the localization of NPY and its receptors, the neuropeptide system has been regarded as the regulating factor in fear learning [39]. Npy knockout mice and Npy1/Npy2 double knockout mice display an impaired ability to extinguish acquired fear memories, accelerated acquisition, enhanced expression/recall, and generalization of fear [40]. Thus, NPY has been regarded as a likely factor underlying pathological fear and a susceptibility factor for PD [41].
Gastrin-releasing peptide (GRP) is a bombesin-like peptide that activates the sympathetic nervous system and has been proposed as a candidate neuropeptide closely associated with fear learning. Grp mRNA is highly expressed in forebrain regions and its receptor shows high immunoreactivity in the amygdala, hippocampus, and several cortical regions in animal models. GRP has thus been regarded as a modulating factor for stress, fear, and anxiety responses. The fear-potentiated startle response, but not anxiety behavior, was specifically blocked by intravenous administration of GRP in rats [42,43]. In addition, mice with knockout of the Grp receptor gene displayed increased and persistent long-term contextual and cue fear conditioning [44,45]. Thus, GRP may be specifically related to anxiety, but not fear. Mice heterozygous for the peptidylglycine α-amidating monooxygenase gene, which is implicated in the activation of neuropeptides, have deficiencies in contextual and cue fear memory [46].
Glucocorticoids
Stress and stress-driven emotional dysregulation are closely associated with the glucocorticoid system and the hypothalamic-pituitary-adrenal axis. Corticotropin-releasing factor (CRF)-immunoreactive cell bodies, terminals, and receptors are specifically located in the basolateral and central nuclei of amygdala. Crf1 and Crf2 knockout mice display absent and attenuated fear responses in the potentiated startle paradigm, respectively [47]. In addition, both susceptibility to PD and childhood risk factors for PD have been associated with single nucleotide polymorphisms in CRF1 receptors and CRF genes [48,49].
Neurotrophins
Mice expressing a common single-nucleotide polymorphism in the brain-derived neurotrophic factor gene (Bdnf, Val-66Met) display increased anxiety-like behavior and impaired fear extinction learning [50,51]. In addition, Bdnf heterozygous mice display impaired contextual fear memory but intact cue fear memory [52]. Furthermore, mice transgenic for the human neurotrophic tyrosine kinase receptor, type 3 (TgNTRK3 mice) display increased panic-like reactions, increased contextual fear conditioning, impaired extinction of contextual fear memories, and exaggerated and pathological fear [17,53]. Based on the observation that tyrosine kinase receptor C (TrkC) is highly expressed in the vicinity of the amygdala-hippocampus-prefrontal fear circuit, and as it is considered a synaptogenic molecule, the exaggerated fear memory in TgNTRK3 mice may be due to hyperexcitability in the CA1 region of the hippocampus [17,54,55]. Greater neuronal activation in the amygdala-hippocampus-prefrontal axis, which results from the impaired extinction of acquired fear, has been observed in TgNTRK3 animals, but not in wild type animals. Thus, TrkC may be regarded as a pivotal protein linking cognitive processes to fear in PD. Anxiety-like behavior and general features of anxiety disorders have been observed in most mouse models with alteration in previously reported PD candidate genes. However, reports of alterations in fear and cognition-related events have been inconsistent [17].
CURRENT PSYCHOPHARMACOLOGICAL INTERVENTIONS AND NOVEL INVESTIGATIONAL THERAPEUTICS FOR PANIC DISORDER
Clinical studies of current pharmacological interventions for PD in the last decade have resulted in strong evidence supporting the effectiveness of paroxetine, venlafaxine, sertraline, fluvoxamine, citalopram, fluoxetine, escitalopram, and clonazepam. In addition, inositol, duloxetine, mirtazapine, milnacipran, and nefazodone have been reported to have anti-panic properties. In addition to selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), TCAs, monoamine oxidase inhibitors (MAOIs), benzodiazepines, and atypical antipsychotics have been proposed as valid alternative interventions for PD. Because of their potential adverse effect profiles, TCAs, MAOIs, benzodiazepines, and atypical antipsychotic have been regarded as not first-line, but second-line, pharmacological intervention options [56].
CBT, pharmacological interventions with antidepressants, and self-help are considered the best treatment options for PD by the National Institute for Health and Clinical Excellence (NICE) guidelines for anxiety disorders [57]. The first choice of pharmacological intervention for PD is the use of an SSRI, and the use of TCAs is permitted. In contrast, the use of benzodiazepines or antipsychotics is not permitted. Benzodiazepine use specifically has been associated with “less good outcome in long term” in the NICE guideline, although relatively favorable safety and effectiveness of benzodiazepines have been demonstrated in recent studies [56]. The use of SNRIs has been not been described in the guideline despite the highquality evidence for its effectiveness in PD [58-60]. Not only SSRIs and TCAs, but also SNRIs and benzodiazepines have been recommended by the American Psychiatric Association practice guideline for the treatment of patients with panic disorder [61]. Moreover, SSRIs (citalopram, escitalopram, fluvoxamine, fluoxetine, paroxetine, and sertraline) and an SNRI (venlafaxine) have been recommended by the World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety and obsessive-compulsive and post-traumatic stress disorders [62]. There is evidence for modest effectiveness of mirtazapine, inositol, milnacipran, nefazodone, and duloxetine, as well as for effectiveness of augmentation of antidepressants with olanzapine, pindolol, aripiprazole, and clonazepam for the treatment of PD in the WFSBP guideline. However, from a clinical perspective, the need for more effective, faster-acting, and more tolerable pharmacological interventions is still unsatisfied. Thus, novel mechanism-based anti-panic therapeutics, including glutamatergic receptor modulators, orexin receptor antagonists, CRF1 receptor antagonists, angiotensin II receptor antagonists, and endocannabinoid system modulators have been proposed [63]. The contents of novel investigational therapeutics for PD are summarized in Table 1.
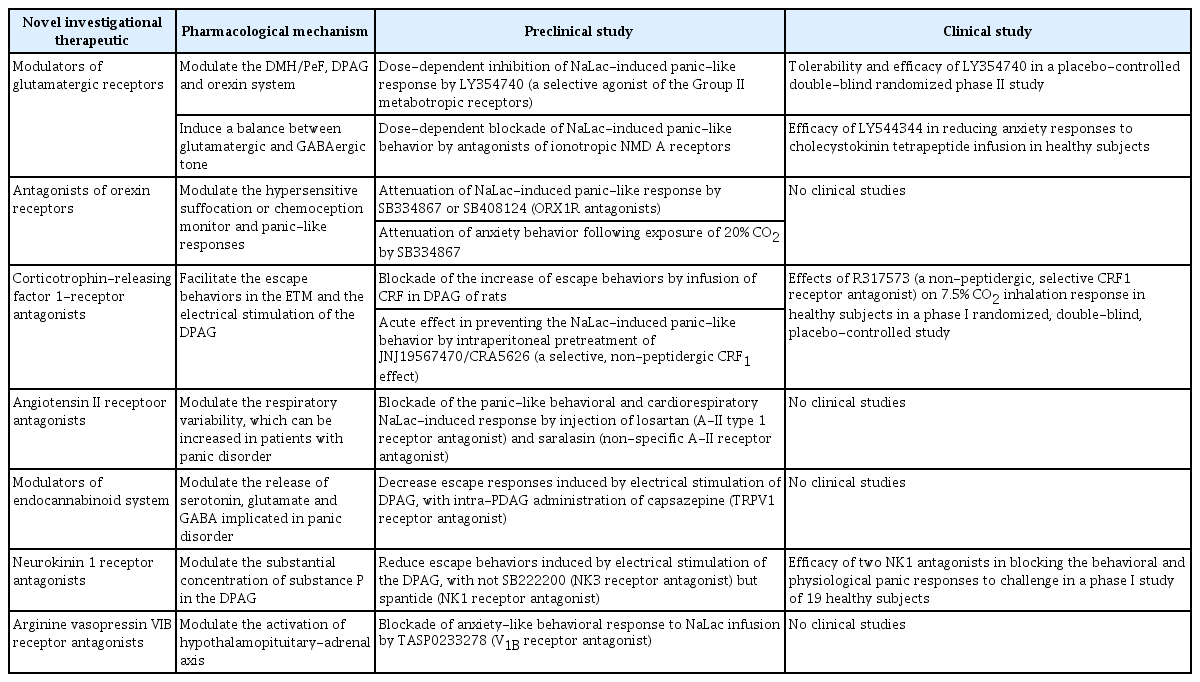
Summary of the novel investigation therapeutics for panic disorder
Glutamatergic receptor modulators
The dorsomedial/perifornical hypothalamic region, dorsal periaqueductal gray, and orexin system, which is implicated in panic/defensive responses, are modulated by glutamate. In addition, imbalance between glutamatergic and GABAergic tone in panic-generating sites has been regarded as a mechanisms underlying panic in a panic-prone rat model [64]. Group II metabotropic glutamate (mGlu) receptors are distributed in the dorsomedial/perifornical hypothalamic region and dorsal periaqueductal gray, which is implicated in panic mechanisms [65]. A dose-dependent inhibition of the sodium lactate (NaLac)-induced panic-like response without sedative effects was observed in panic-prone rat model following pretreatment with LY354740 (selective agonist of Group II mGluRs) [66]. In addition, NaLac-induced panic-like responses were blocked in a panic-prone rat model following pretreatment with Cbi-PES and THIC, which are selective agonists of Group II mGlu receptor type 2 (mGluR2). Therefore, mGluR2 agonists have been proposed as promising anti-panic drugs [65]. The tolerability and efficacy of LY354740 (4 weeks of treatment, 200 mg) were studied in a placebo-controlled double-blind randomized phase II study in subjects subjected to panic attacks induced by a single inhalation of a mixture of 35% CO2 and 65% O2. Although LY354740 was shown to be superior to the placebo in reducing CO2-induced panic attacks after 4 weeks of treatment and led to subjective clinical improvements, the findings of the study were not replicated in another placebo-controlled randomized double-blind phase II study [67]. The efficacies of LY354740 (100 to 200 mg), paroxetine (active comparator) (40 mg), and placebo were compared over a course of 9 weeks. No significant difference was observed between LY354740 and placebo. The lack of efficacy of LY354740 may be due to its poor oral bioavailability [68]. The efficacy of the pro-drug LY544344 in reducing anxiety responses to cholecystokinin tetrapeptide (CCK-4) infusion was investigated in a preliminary phase I study, although the CCK-4 challenge test has not been clearly validated as an experimental model of panic attacks [69].
In addition, injection of an N-methyl-D-aspartate (NMDA) receptor antagonist (MK-801) into the dorsomedial/perifornical hypothalamic region led to a dose-dependent blockade of NaLac-induced panic-like behavior. In contrast, injection of GYKI52466, which does not affect NMDA receptors, into the dorsomedial/perifornical hypothalamic region did not prevent panic responses [70]. Thus, post-synaptic mechanisms involving NMDA-glutamate receptors have been proposed as potential contributors to panic vulnerability.
Orexin receptor antagonists
The orexin (ORX) system projects to the dorsal periaqueductal gray and limbic system, and is implicated in defense and emotional responses. The ORX system has been proposed as a key substrate implicated in the “suffocation/chemoception monitor” and panic-like responses [71]. Panic-like responses were shown to be attenuated without sedative effects in panicprone rats following pretreatment with SB334867 or SB408124 (ORX1 receptor antagonists) [72]. In addition, pretreatment with SB334867 has been shown to attenuate anxiety behavior following exposure to 20% CO2 [73]. Because they reduce hyperreactivity in response to panicogenic stimuli, ORX1 receptor antagonists have been proposed as a potential novel approach for the treatment of PD.
Corticotrophin-releasing factor 1 receptor antagonists
Pretreatment with antalarmin (CRF1 receptor antagonist) was shown to block increased escape behaviors induced by infusion of CRF into the dorsal periaqueductal gray in rats [74]. In addition, intraperitoneal pretreatment with JNJ19567470/CRA5626 (CRF1 receptor antagonist) prevented NaLac-induced panic-like behaviors and cardiovascular responses without sedative effects [75]. In a phase I randomized, double-blind, placebo-controlled study, treatment with R317573 (CRF1 receptor antagonist) (40 mg, 7-day treatment) significantly reduced CO2-induced behavioral responses without sedative effects [76]. However, due to the arbitrary nature of the measurement of significant responses to inhalation, there has been no conclusion regarding the specific anti-panic properties of R317573.
Angiotensin II receptor antagonists
Neurons producing angiotensin II (A-II) are located in circumventricular organs, which are connected to the dorsomedial/perifornical hypothalamic region in panic-prone rats [77]. A-II has thus been proposed as a potential modulator of respiratory variability in PD. Injections of losartan (A-II type 1 receptor antagonist) and saralasin (non-specific A-II receptor antagonist) into the dorsomedial hypothalamic regions of panic-prone rats has been shown to block panic-like behavioral and cardio-respiratory NaLac-induced responses [78].
Endocannabinoid system modulators
Cannabinoids modulate the release of several neurotransmitters involved in panic mechanisms, including serotonin, glutamate, and GABA. Intra-dorsal periaqueductal gray administration of capsazepine [transient receptor potential vanilloid type-1 (TRPV1) receptor antagonist] was shown to decrease escape responses induced by electrical stimulation of the dorsal periaqueductal gray [79]. Furthermore, inhibition of anandamide (AEA) using the enzyme fatty acid amide hydrolase (FAAH) increases endocannabinoid system activity. Thus, N-arachidonoyl-serotonin (dual FAAH/TRPV1 blocker), which inhibits AEA degradation and is a TRPV1 receptor antagonist, has been proposed as a potential treatment for a model of general anxiety [80,81].
Neurokinin 1 receptor antagonists
Neurokinin (NK) and substance P (SP) receptors has been proposed to have potential roles in panic attacks, as NK receptors (NK1, NK2, and NK3) are distributed in brain regions involved in emotion/anxiety and memory/learning, and substantial concentrations of SP are present in the dorsal periaqueductal gray [82,83]. Treatment with spantide (NK1 receptor antagonist), but not SB222200 (NK3 receptor antagonist), decreased escape behaviors in panic-prone rats. In a randomized, double-blind, placebo-controlled phase I study, a single oral dose of vestipitant (GW597599) (15 mg), a single intravenous dose of vofopitant (Gr205171), or a single oral dose of alprazolam (0.75 mg) was administered to 19 healthy subjects prior to a 7% CO2 challenge. Vestipitant was shown to have anxiolytic effects, while vofopitant did not demonstrate any panicolytic effects. It has been speculated that the lack of panicolytic effects of vofopitant may be due to the high variability in its pharmacokinetic coefficients. Thus, vestipitant (NK1 antagonist) has been proposed to have potential efficacy in preventing the behavioral component of panic attacks [82].
Arginine vasopressin V1B receptor antagonists
Arginine vasopressin (AVP) is a cyclic nonapeptide involved in the activation of the hypothalamic-pituitary-adrenal axis. Of the AVP receptors (V1A, V1B, and V2), the V1B receptor is mainly distributed in pituitary neurons. Pretreatment with TASP0233278 (V1B receptor antagonist) has been shown to block NaLac-induced anxiety-like behavioral responses in panic-prone rats. However, the NaLac-induced respiratory response was not altered by TASP0233278 [83]. Therefore, the modulating effects of V1B receptor antagonists on the hypothalamic-pituitary-adrenal axis are considered not crucial for the prevention of panic attacks.
Despite the small amount of progress in studies of alternative novel investigational therapeutics, glutamate- and ORX-related molecules are considered the most promising potential treatments for PD. mGluR2 receptor agonists and ORX1R antagonists have been shown to block panic-like responses without sedative effects in preclinical studies. However, clinical studies of the effectiveness of glutamate- and ORX-related novel therapeutics have been inconclusive. Further clinical trials are required in addition to the use of robust neurobiological theoretical foundations and promising preclinical data regarding panic mechanisms [63].
NEUROANATOMICAL CORRELATES OF PANIC DISORDER
Reductions in cortico-limbic structure volumes have been mainly associated with panic symptoms, whose maintenance might be due to various ontogenetic disturbances. The hyperexcitability of neurotransmission may be associated with a reduction in amygdala volume, and amygdala activation may also contribute to increased brainstem and insula volume. In addition, it has been speculated that abnormalities in hippocampal and parahippocampal areas in the resting state are associated with the bidirectional connection between the hippocampus and amygdala implicated in the mediation of PD-related traits. Furthermore, involvement of the amygdala in spontaneous panic attacks may be associated with the assumption that the amygdala may be the core structure within the fear network in the initiation of panic attacks. The amygdala-hippocampus-prefrontal cortex axis has been regarded as the essential neuroanatomical correlate of PD. The association between each of the above brain regions and fear regulation in PD is discussed below [17,18,84].
Amygdala
Reductions in amygdala volume in patients with PD have been consistently reported in structural magnetic resonance imaging (sMRI) studies [85-88]. In addition, a functional MRI (fMRI) study reported decreased amygdala activation upon the viewing of fearful faces in patients with PD, but not in healthy controls [89-91]. This observation may be explained by the basal activation-induced reduced emotional response to fear-related stimuli. Moreover, increased amygdala activation in emotional conflict paradigms, such as the Stroop-like presentation of incongruent and congruent emotional stimuli, has been reported in an fMRI study and may be explained by abnormal cognitive information processing at high-level centers [17,18].
Hippocampus
Several sMRI studies have reported inconsistent findings regarding hippocampal volume changes in patients with PD. Both of hippocampal volume reductions and increases have been reported in patients in PD, but not in healthy controls. In addition, no differences in hippocampal volume between patients with PD and healthy controls have been reported [86-88,92,93]. Hippocampal volume variations in patients with PD may be due to the influences of confounding factors. First, antidepressant-induced hippocampal neuroplasticity can influence the findings of sMRI studies, as antidepressant treatment cannot be excluded in clinical studies of patients with PD for ethical reasons [94-96]. Second, variations in the findings may be due to variations in the levels of lifelong stressors experienced by the individual, as stress-associated hippocampal neuronal damage has been observed in animal models [88]. Thus, the reduction in hippocampal volume has been reported to be more prominent in the early phase of PD rather than in the late phase, as hippocampal volume changes in the late phase are masked by the potential effects of antidepressant treatment or lifelong stressors. Hippocampal neuronal damage has been reported in a spectroscopic study, as indicated by alterations in the ratio of N-acetylaspartate to creatine in the left hippocampus [97]. Moreover, functional alterations in benzodiazepine receptor binding comprising decreased benzodiazepine receptor binding in the left hippocampus and increased benzodiazepine receptor binding in the hippocampus/parahippocampal gyrus have been reported [98]. A specific right-left asymmetry in the parahippocampal gyrus in patients with PD, but not in healthy controls, has been reported in a PET study of a panic-provoking agent including sodium lactate. In addition, a genetic mouse model transgenic for the human neurotrophic tyrosine kinase receptor, type 3 was shown to have increased fear memory and hyperexcitability in the CA1 sub-region of the hippocampus. This model also displayed abnormalities in the amygdala-hippocampus-prefrontal cortex fear circuit in fear conditioning and fear extinction paradigms [17,18].
Prefrontal cortex
The anterior cingulate cortex is a prefrontal region implicated in the pathogenesis of PD. Reduction in anterior cingulate cortex volume has been reported in an sMRI study of patients with PD [85,99]. Increased anterior cingulate cortex activation in response to happy faces and decreased anterior cingulate cortex activation in response to fearful faces has been reported in an fMRI studies of patients with PD [90,91]. Reduced activation of the subgenual cingulate in response to threats and increased sensitivity of this region to safe conditions was reported in patients with PD during an instructed fear-conditioning paradigm [100]. Thus, it may be speculated that the generalized activation of the anterior cingulate cortex in response to neutral/safe stimuli is attributed to chronic hyperarousal in patients with PD. In line with this speculation, patients with PD have been shown to have deficits in discrimination learning, as indicated by presentation of an enhanced startle potentiation response to learned safety rather than abnormal reactivity to a danger cue [101]. In contrast, the anterior cingulate cortex has been shown to have hypoactive responses during the processing of emotionally negative or conflictive information. This response is associated with the translation into disinhibition in the amygdala and an increase in fear expression in patients with PD [102].
The orbitofrontal cortex has also is also regarded as a potential areas of the medial frontal cortex with abnormal functioning in PD [103,104]. Reduction of in the volume of the gray matter in the orbitofrontal cortex in patients with PD, but not in healthy controls, has been reported in a number of studies [93,105-107]. In addition, decreased baseline cerebral blood flow in the orbitofrontal cortex has been shown to be related to the potential prediction of panic vulnerability and is inversely associated with anxiety scores in patients with PD [105]. Moreover, although the insular cortex has been proposed as an area potentially related to the misinterpretation of bodily sensations and biased cognitive evaluation of stimulus valence in PD, reports of gray matter volume changes in this region have been inconsistent. Despite the inconsistent conclusions, altered GABA receptor binding has been reported in insular regions in patients with PD [108-110].
Several aspects of the neuroanatomical correlates of PD should be considered. First, findings of structural and functional alterations in PD have been inconsistent. In addition, disturbances in control of limbic structures by the prefrontal cortex and anterior cingulate cortex cannot sufficiently explain the pathogenesis of PD. The fear circuit, which includes the amygdala-hippocampus-prefrontal cortex axis, should have more sophisticated specific characteristics that may be explained based on various influential factors. Second, complex human-specific processes in PD might be concealed in the derivation of neuroanatomical models based on animal research. In order to better understand the complex neuroanatomical underpinnings of PD pathogenesis, the model used to explain the functions of the prefrontal cortex should be more intricate. Third, information regarding neurochemical alterations in PD should be coupled with those regarding the fear circuit. Thus, alterations in GABAergic transmission in fronto-temporal areas and changes in serotonergic transmission in dorsal raphe nuclei may contribute to the creation of more sophisticated models of fear circuits in PD. Fourth, it is expected that refinement of distorted emotional processing in PD can be supported by conceptualizing genetically driven dysfunctional cortico-limbic interactions. Therefore, to guide future decisions based on neuroanatomy, more comprehensive modeling of the neuroanatomical network underlying PD can be supported by potential genetic factors shaping neural network activity [17,18].
NEURAL CORRELATES OF COGNITIVE-BEHAVIORAL THERAPY FOR PANIC DISORDER
The efficacy and effectiveness of CBT for PD have been supported by several meta-analytic reviews. Since the neural correlates of CBT for PD have only been investigated in two PET studies, one single photon emission computed tomography study, and one fMRI study, the neural mechanisms underlying the effects of CBT have remained largely unknown [111-116]. The “PANIC-NET” multicenter research project was conducted to investigate CBT for PD in large-scale randomized controlled trials from 2006 to 2013. Treatment with two variations of CBT, therapist-guided and self-guided exposure, was administered to 369 medication-free patients with PD. Experimental fMRI and genotyping add-on studies were performed before and after CBT [117]. In the “PANIC-NET” multicenter study, the influence of CBT on the neural correlates of fear conditioning was investigated in 42 patients with PD. The increased inferior frontal gyrus activation before therapy in patients with PD was normalized by CBT. In addition, there was a correlation between reduced activation in the inferior frontal gyrus and the reduction in agoraphobic symptoms from baseline to the post-measurement time point [115,118]. These findings support the idea that altered top-down cognitive processing, including increased threat expectancy and attention to threat, is normalized by CBT. The connectivity between the inferior frontal gyrus and the fear network (amygdala, hippocampus, anterior cingulate cortex, and medial prefrontal cortex) in patients with PD has been reported to be increased during CBT. It has been speculated that changes in fear network processing (reduced top-down processes) might be caused by the normalization of inferior frontal gyrus activation during CBT [119,120].
The “PANIC-NET” multicenter study indicated that non-responders, rather than responders, displayed enhanced activation of a network including the pregenual anterior cingulate cortex, amygdala, and hippocampus during safety signal processing prior to treatment onset. There was also an association between treatment response and enhanced negative functional coupling of the anterior cingulate cortex and amygdala. The inhibitory top-down modulation of the amygdala via medial prefrontal cortex inputs and functional connectivity between the amygdala and the ventro-medial prefrontal cortex may thus be the neural correlates of successful fear extinction. It has been speculated that patients with PD with relatively strong inhibitory coupling between these structures prior to therapy have an increased chance of benefitting from exposure in situ [111]. The “PANIC-NET” multicenter study investigated the impact of MAOA promoter-associated variable number tandem repeat polymorphisms (MAOA-uVNTRs) on therapy response. Worse outcomes, as measured using the Hamilton Anxiety Rating Scale, were significantly associated with the presence of long MAOA-uVNTR alleles in patients with PD [121,122].
DISCUSSION
Further integration of behavioral, psychophysiological, neurobiological, and genetic data is needed to validate the pathways of therapeutic change in CBT and pharmacotherapy for PD. The aims of pharmacotherapy for PD may be to improve learning during CBT, which targets plasticity in cortical structures and has been proven to be effective in both rodents and humans. The use of pharmacotherapy together with D-cycloserine resulted in reductions in major acrophobic symptoms in a successful example of combination therapy [17,18,123].
PD is modeled in terms of the negative valence systems domain in the Research Domain Criteria (RDoC) initiative. A panic attack (Criteria A) can be conceptualized as a prototypical expression of a fear response to an acute internal threat stimulus, while concerns and worries about the consequences of panic attacks (Criteria B) can be conceptualized as responses to potential harm within the negative valence system. Thus, within the negative valence systems domain, PD can fit into the threat imminence model. The contents of the model are as follows: perceived proximity and imminence might contribute to the specific types of defensive behaviors. In an animal model, threat-nonspecific vigilance and other preemptive behaviors can be engaged in the context of firstly encountered threat in terms of pre-encounter defense. In contrast, fear bradycardia, amygdala-dependent potentiation of the startle reflex, and amygdala-to-periaqueductal gray matter projection-dependent motor freezing may result from selectively allocated attention to the threat cue in terms of a post-encounter defense. Active defense behaviors, including active avoidance and flight or fight responses, can be initiated by mediation of the dorsal periaqueductal gray and accompaniment of a general discharge of the sympathetic nervous system in the context of a circa strike defense. Considering a prior panic attack as an undetected body symptom, preemptive behaviors including safety behaviors and threat-nonspecific hypervigilance can be considered pre-encounter defense. In contrast, anxious apprehension can be considered post-encounter defense. The above model is supported by the finding of a clear potentiation of the startle response during recovery from hyperventilation in patients with PD, but not in healthy controls. Based on the proposal by Craske, a panic attack may be considered an example of circa strike defense. The proposal is supported by the findings that active avoidant behavior is facilitated by potassium cyanide-induced hypoxia in the dorsal periaqueductal gray, and that potassium cyanide-evoked escape behavior is abolished by lesions in the dorsal periaqueductal gray [14,124-126].
An essential component of “personalized medicine” is the use of reliable indicators of predictions/outcomes, which are used to choose the most appropriate therapeutic for subpopulations of patients with PD with specific neurobiological and symptomatological features. Consistent with the idea of “personalized medicine”, the RDoC aims to create a shift from existing diagnostic categories to observable behavioral and neurobiological measures. In the RDoC, the “negative valence systems” domain, which includes fear and anxiety, is structured around “units of analysis” of genes, molecules, cells, brain circuits, and behaviors. In addition, the definitions of different subtypes of PD may benefit from the use of the RDoC. The RDoC strategy is thought to contribute to further treatments targeting the underlying dysfunctions in PD more precisely [14,17,18]. It can also be used to define moderators to identify subgroups based on differences in response to treatment. Therefore, in the novel bio-psychosocial-behavioral treatment model of PD, glutamate- and ORX-related molecular mechanisms associated with the fear circuit including amygdala-hippocampus-prefrontal cortex may need to be targeted.