The Mechanism of Anti-Epileptogenesis by Levetiracetam Treatment is Similar to the Spontaneous Recovery of Idiopathic Generalized Epilepsy during Adolescence
Article information
Abstract
Objective
The anti-epileptogenic drug levetiracetam has anticonvulsant and anti-epileptogenesis effects. Synergy between cell death and inflammation can lead to increased levels of apoptosis inhibitory factors and brain-derived neurotrophic factor, aberrant neurogenesis and extended axon sprouting. Once hyperexcitation of the neural network occurs, spontaneous seizures or epileptogenesis develops. This study investigated whether the anti-epileptogenic effect of levetiracetam is due to its alternate apoptotic activity.
Methods
Adult male Noda epileptic rats were treated with levetiracetam or vehicle control for two weeks. mRNA quantification of Bax, Bcl-2 and GAPDH expression were performed from prefrontal cortex and hippocampus tissue samples.
Results
The levetiracetam-treated group showed a significant increase of Bax/Bcl-2 mRNA expression ratio in the prefrontal cortex than the control group, but no change in the Bax/Bcl-2 mRNA expression ratio in hippocampus.
Conclusion
Idiopathic generalized epilepsy including childhood absence epilepsy develop at childhood and recover spontaneously during adolescence. The aberrant neural excitable network is pruned by a neural-maturing action. This study suggests the mechanism of acquired anti-epileptogenesis by levetiracetam treatment may be similar to spontaneous recovery of idiopathic generalized epilepsy during adolescence.
Introduction
Idiopathic generalized epilepsy, such as childhood absence epilepsy, develop at childhood and recover spontaneously during puberty, patients with other types of epilepsy are often prescribed medicine throughout their lifetime. Available anticonvulsant drugs may prevent seizures, but do not cure epileptogenesis.12 The antiepileptic action of most anticonvulsants acts on ion-channels. Many ion-channel related genes have been identified as susceptibility genes for epilepsy.3 Although seizures are inhibited during anticonvulsant treatment, they can occur again following cessation of treatment. Therefore, the action of ion-channels is related to the seizure, but not to epileptogenesis itself.4 It was previously suggested that regulation of ion-channels and neurotransmitters involved an anti-ictogenic action, whereas neurotransmitters, neurotrophins, apoptosis, or nitric oxide synthase may contribute to the anti-epileptogenic effect.4 In temporal lobe epilepsy (TLE), a type of symptomatic localization-related epilepsy, evidence increasingly suggests that changes in the neural network are involved in acquiring epileptogenesis because of the limited focus region in the inner surface of the temporal lobe, such as the hippocampus. Decreased volume of the hippocampus due to neuronal loss is known as hippocampal sclerosis. In addition, the neurogenesis of ectopic granular cells occurs concurrently with neuronal death, and aberrant axonal sprouting (mossy fibers) are observed in the brain.5 The principal mechanism of epileptogenesis is due to the excitability of the neural network, both by ectopic neurons or axons.56 The Bcl-family, known modulators of apoptosis, and neurotrophins may be related to aberrant neurogenesis and extended axons. Indeed, apoptosis inhibitory factors such as brain-derived neurotrophic factor (BDNF),7 Bcl-2 and Bcl-XL,8 were increased in the postmortem brain of patients with TLE associated with hippocampal sclerosis.
Symptomatic localization-related epilepsy can develop after the occurrence of brain infarction, head trauma, head surgery, Alzheimer's disease, or encephalitis. During brain infarction or head trauma, injured neurons secrete adenosine triphosphate (ATP). Discharged ATP subsequently activates microglia, cells which monitor the behavior of neurons, which then secrete inflammatory cytokines, such as IL-1β, which can induce an inflammatory repair response.910 In Alzheimer's disease, accumulated amyloid-β can behave as an inflammasome,11 and induce inflammation as an innate immune defense of the brain.1213 In addition, increased inflammatory mediators such as IL-1β are observed in Rasmussen encephalitis. Moreover, intractable seizures can be cured by treatment with ACTH, steroids, immunoglobulins, plasmapheresis and immunosuppressants, suggesting a relationship between inflammation, seizures, and epileptogenesis.14 Animal models of epilepsy are induced by injecting kainic acid into the brain. Kainic acid induces cell death, and the subsequent release of ATP from dead cells activates microglia to produce IL-1β that activates astrocytes. Alpha-2 macroglobulin secreted from activated astrocytes induces the extension of neuronal axons, and repairs damaged neurons. In addition, apoptosis may promote neurogenesis.15 The EL mouse,16 a natural model of epilepsy, develops epileptogenesis with spontaneous seizures from 10 weeks of age. Apoptosis related factors, neurotrophin,17 inflammatory cytokines18 and cell cycle components19 are significantly altered at the time of epileptogenesis. In addition, Bcl-2, and BDNF levels are increased. However, cytokine levels return to the baseline, and cell cycling is normalized at 24 weeks of age in the EL mouse.
Synergy between cell death and inflammation increases levels of apoptosis inhibitory factors and BDNF, induces aberrant neurogenesis and extension of axons, and eventually leads to a hyper excitable neural network which causes epileptogenesis. Previous studies suggest that epileptic seizures promote apoptosis and increase BDNF levels.20 Thus these sensitization phenomena may promote epileptogenesis. Functional genomic analysis by microarray demonstrated significant changes in gene expression related to cell death, acute inflammatory responses, and synaptic vesicles.21 Synaptic Vesicle 2A (SV2A) is a component factor of the synaptic vesicle, and exocytosis of neurotransmitters from vesicles is decreased in SV2A deficient mice.22 It has been reported that SV2A is decreased both in postmortem brains with TLE23 and epilepsy-associated brain tumors.24
Levetiracetam is a pyrrolidone derivative, and used as an anticonvulsant,25 which does not mediate its effects through neurotransmitters (glutaminergic acid, GABA, dopamine, serotonin) or ion-channel proteins,26 but is linked to SV2A.27 Since the efficacy of levetiracetam is decreased in SV2A-deficient mice, it is likely that the clinical efficacy of this compound is related to the modulation of neurotransmitters via SV2A.22 Levetiracetam has been shown to have both anticonvulsant efficacy and antiepileptogenic action in animal models, such as the spontaneous epileptic rat (SER).2 SER was established as a double mutant by crossing tremor rats and zitter rats. SER were pretreated with levetiracetam before developing epileptogenesis to maintain anticonvulsant efficacy when stopping levetiracetam treatment, as seizures have been observed immediately after discontinuing treatment with other anticonvulsants,28 In addition, pre-treatment may decrease the number and duration of seizures, even after stopping the levetiracetam treatment.29 It has been reported that the progression of kindling was markedly delayed in levetiracetam-pretreated amygdala kindling rats due to the inhibition of increasing levels of BDNF.30 It may be predicted that Bcl-2 expression is also inhibited if levetiracetam inhibits BDNF, as the antiepileptogenic action of levetiracetam is due to the decreased expression of BDNF and Bcl-2. However, as the ratio of Bax/Bcl-2 expression levels is an indispensable biomarker for the inhibition of apoptosis, the primary endpoint for the current study was the ratio of Bax/Bcl-2 mRNA expression in the prefrontal cortex and hippocampus after levetiracetam treatment of Noda epileptic rats (NER). NER is a natural model of Epilepsy, spontaneous convulsions are controlled by autosomal recessive gene for epilepsy, therefore, they are comparable to generalized epilepsy for humans.31
METHODS
Animals and drug administration procedure
Adult male Noda epileptic rats aged 5 weeks were housed under a 12-h light-dark cycle at a temperature of 19–20℃ with free access to both food and water. All procedures involving animals were performed in accordance with Animal Care Guidelines and approved by the Osaka Medical College Committee on Animal Care and Supply. After 1 week of acclimatization, rats were divided into two groups (n=6 per group). The rats were given daily inter-peritoneal (i.p.) drug injections for two weeks. Group 1 received levetiracetam (80 mg/kg) and group 2 received a vehicle control (0.9% saline).
RNA isolation and cDNA synthesis
Brains were dissected and placed on an ice-cold plate. The samples from the prefrontal cortex and hippocampus were rapidly dissected and immediately frozen in liquid nitrogen and stored at −80℃. Total RNAs were isolated from the tissues using a Qiagen RNeasy Minikit (Qiagen, Hilden, Germany), and subjected to first-strand cDNA synthesis with the reverse transcriptase (RT) OMNISCRIPT (Qiagen), using a random primer (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. A total RNA of 200ng (based on absorbance) was transcribed into cDNA and added to each reaction of the Quantitative RT-PCR assay.
Primers and probes
Quantification of the mRNA expression for Bax, Bcl-2 and GAPDH were performed using the LightCycler apparatus (Roche Diagnostics, Basel, Switzerland). Primers and probes used were designed with the assistance of Oligo 4.0 (National Biosciences, Plymouth, MN, USA) and synthesized by Nihon-Idenshiken (Sendai, Japan).
Construction of the plasmid calibrators
The transcripts that encode for Bcl-2, Bax and GAPDH were amplified by RT-PCR using specific primers. The PCR product of each gene was cloned into the pGEM T easy Vector (Promega, Madison, WI, USA) and sequenced. The standard curves were generated from serially diluted solutions of each plasmid clone as templates.
Data analysis
Statistical comparison of data from levetiracetam and sa-line-treated groups was performed using a two-tailed Student's t-test. A value of p<0.05 was considered statistically significant.
RESULTS
Primary endpoint
The Levetiracetam-treated group showed a significant increase in Bax/Bcl-2 mRNA expression ratios from the prefrontal cortex compared with the control group (Mean±SD; 1.40±0.31 vs. 0.96±0.14 respectively, p=0.02) (Figure 1). However, levetiracetam treatment did not alter the Bax/Bcl-2 mRNA expression ratio in the hippocampus compared with the control group (0.78±0.17 vs. 0.80±0.27 respectively, p=0.90).

Expression ratio of Bax/Bcl-2 mRNA (primary endpoint). A: Prefrontal cortex. B: Hippocampus.
Secondary endpoint
The levetiracetam-treated group showed an increase in Bcl-2 mRNA expression levels in the prefrontal cortex compared with the control group (relative values when the expression of GAPDH mRNA is regarded as 1,000; 8.7±2.5 vs. 6.2±1.8 respectively, p=0.08) (Figure 2). Levetiracetam treatment did not alter Bax mRNA expression levels compared to the control group (8.2±0.8 vs. 8.1±1.0 respectively, p=1.00). In the hippocampus there was no differences in the mRNA expression level of Bax and Bcl-2 between the levetiracetam-treated and control groups (Bcl-2: 15.0±4.3 vs. 14.6±5.2 respectively, p=0.90; Bax: 11.2±2.0 vs. 10.6±0.9; respectively, p=0.48).

Expression level of Bax or Bcl-2 mRNA (secondary endpoint). A: Bcl-2 mRNA expression level in prefrontal cortex. B: Bax mRNA expression level in prefrontal cortex. C: Bcl-2 mRNA expression level in hippocampus. D: Bax mRNA expression level in hippocampus.
DISCUSSION
The current study observed increased Bax/Bcl-2 ratios at the prefrontal cortex in Noda epileptic rat, which likely contribute to a decrease of Bcl-2 rather than an increase in Bax levels. However, this association was only evaluated by analyzing mRNA expression of Bax and Bcl-2, whereas down-stream apoptosis exective factors such as caspases were not analyzed. In addition, we did not perform nuclear fragmentation analysis, a marker for apoptotic cell death, or monitor behavioral influences. Although our current findings should be regarded as preliminary, further study is required to determine the relationship between altered mRNA levels, expression of proteins, and fragmentation of the nucleus as apoptosis regulating/performance factors with the inhibition of seizures or anti-epileptogenesis effect. Furthermore, the Bax/Bcl-2 ratio was up-regulated in the prefrontal cortex of levetiracetam-treated rats, whereas it was not observed in the hippocampus. Compared with the Wistar rat, SV2A expression in the parietal-temporal cortex was significantly increased in Noda epileptic rats,32 but not in the hippocampus. Levetiracetam is reported to have a diminished clinical efficacy on decreasing SV2A expression. Thus, levetiracetam may not have an effect on the apoptosis regulating factor in Noda epileptic rat hippocampus, as the SV2A expression level is same as normal rats. Although the volume and duration of levetiracetam-treatment was determined according to a previous study,28 the anti-epileptogenesis effect of levetiracetam may be different in other strains of rat, and therefore the volume and duration used in this study were not optimized for the Noda epileptic rat. The transition of Bax/Bcl-2 expression required to induce anti-epileptogenesis in the EL mouse was previously reported, and the expression pattern differed between the frontal lobe cortex and hippocampus.17 Further analysis is needed to assess the expression pattern of Bax, Bcl-2, Bcl-XL, and other apoptosis-regulating factors throughout the duration of the treatment.
The anti-epileptogenesis mechanism of levetiracetam
Focal cortical dysplasia is a brain malformation with hyperplasia in the cortex, which induces intractable epilepsy. The mechanism of cortex hyperplasia involves the inhibition of apoptosis, increased BDNF levels, and the formation of an aberrant neuronal network. Assessment of dysplastic cells from cortical dysplasia patients demonstrated that the expression of the NR2A subunit was decreased to a quarter of the control level, whereas the NR2B subunit was increased ten times, and therefore the NR2A/NR2B ratio was 1:40.33
The NMDA receptor expression pattern is different according to the developmental stage of the brain. The main subunits are NR2A and NR2B, and the NR2A/NR2B ratio is low in in-fants and higher in adolescent years, whereas it becomes lower than adolescent at young adult years on human postmortem brain.34 The NR2A subunit does not have a nitric oxide synthase (NOS)-binding site, although it is present in NR2B. NOS produces nitric oxids (NO), a type of gas, and therefore it can circulate easily throughout the brain and activate guanylate cyclase (GC) present in the surrounding brain cells.
The activity of GC is 50 times higher in NR2B than NR2A. GC is an enzyme which mediates GTP to cGMP transformation. cGMP activates protein kinase G (PKG), by phosphorylating Ser9 in glycogen synthase kinase-3 beta (GSK-3β) which inhibits activation of GSK-3β. GSK-3β is involved neuroprotective effect in the human brain, and its inhibition allows in-creased levels of BDNF and Bcl-2, which lead to inhibition of apoptosis, and the start of neurogenesis. Hyperplasia in the brain cortex during focal cortical dysplasia may be due to the excessive neuroprotective effect afforded by the diminished NR2A/NR2B ratio.33 While the lower NR2A/NR2B ratio in infants is neuroprotective, the higher ratio in adolescents is regarded as neuropathic. The realignment of the immature brain is thought to be due to the pruning of unnecessary neural networks, to ensure the mature brain consists of only high-quality neurons. Therefore, the higher NR2A/NR2B ratio in adolescents is considered a neuron-maturing effect.
With idiopathic generalized epilepsy, such as childhood absence epilepsy, the illness spontaneously recovers during puberty. This may be due to an increased NR2A/NR2B ratio, which directs aberrant excitable neural networks to be pruned at that time. Neuroprotection or neuron-maturing is determined by the activation or suppression of GSK-3β. GSK-3β is neuroprotective when suppressed, but neuron-maturing when activated. GSK-3β is activated following phosphorylation at Tyr 216, or suppressed by phosphorylation at Ser 9. The default status of GSK-3β is activated. Without stimulation, or with an inappropriate input into the neuron, GSK-3β remains activated by autophosphorylation, and therefore the neuron will undergo apoptosis or sub-lethal apoptosis.35 Conversely, the upstream kinase of GSK-3β can phosphorylate Ser9 and upon the appropriate input, the neuron will survive. The upstream cascade of GSK-3β contains cytokine pathways, G protein-coupled receptors (GPCR), ion channels, and the Wnt pathway (Figure 3). Various input stimuli can activate GSK-3β and induce cell death. However, the range between appropriate or inappropriate input stimuli is different between diseases or ages (Figure 4).

The intracellular signal transduction course which it is assumed to be associated with SV2A accommodation and nervous system maturity action of Levetiracetam. Arrow: promotion, T shape bar: inhibition, Red color: activated, Blue color: hypofunction. AC: adenylyl cyclase, ATP: adenosine triphosphate, BDNF: brain-derived neurotrophic factor, cAMP: cyclic adenosine monophosphate, CaM: calmodulin, cGMP: cyclic guanosine monophosphate, CN: calcineurin, CREB: cAMP response element binding protein, Cyt C: cytochrome c, DISC1: disrupted schizophrenia 1, DVL: mammalian homologue of drosophila dishevelled, EGF: epidermal growth factor, GC: guanylyl cyclase, GTP: guanosine triphosphate, FZD: frizzled, GSK3β: glycogen synthase kinase 3β, GPCR: G protein-coupled receptor, IL-2: interleukin 2, LRP: LDL-receptor-related protein, NFAT: nuclear factor of activated T cells, NO: nitric oxidase, NOS: nitric oxidase synthase, PDE: phospho-di-esterase, PDK1: phosphatidylinositide-dependent kinase 1, PD-1: programmed deth 1, PI3K: phosphatidylinositol 3 kinase, PKA: protein kinase A, PKC: protein kinase C, PKG: protein kinase G, PLC: phospholipase C, TCF: T-cell factor.
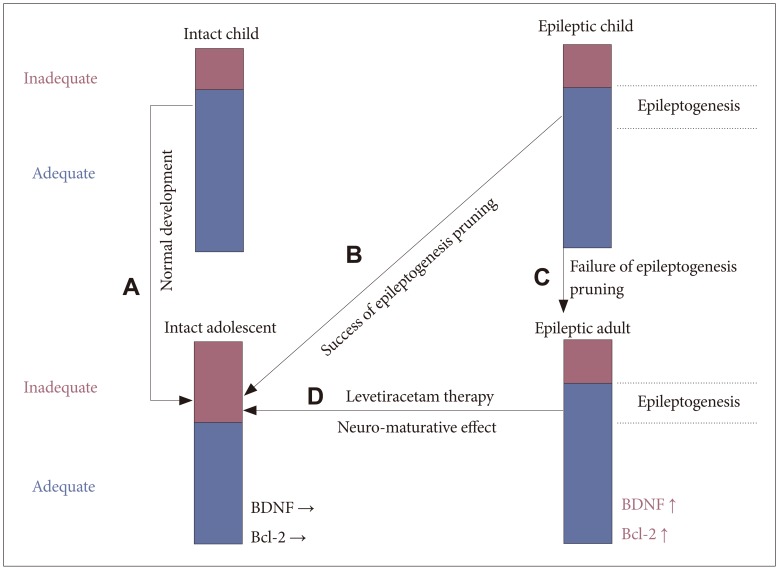
Hypothesis: the mechanism of anti-epileptogenesis of levetiracetam is same as the spontaneous recovery of idiopathic generalized epilepsy during the adolescent period.
During infancy, the range of input stimuli is judged as appropriate for neuroprotective efficiency due to the high NR2A/NR2B ratio. Conversely, neuron-maturing efficiency is increased during adolescence due to the changed range to judge the input stimuli as inappropriate (Figure 4A). This may suggest why idiopathic generalized epilepsy spontaneously resolves during the realignment of the adolescent brain network. Simply, the aberrant neural excitable network is pruned by a neural-maturing action (Figure 4B). When spontaneous recovery does not occur, it is likely that the neuron-maturing action has not been activated (Figure 4C). The NR2A/NR2B ratio decreases again after adolescence and therefore idiopathic generalized epilepsy is not easily cured in adults if the aberrant neural excitable network is not pruned at adolescence.
Further discussion is warranted for the relationship between neurotransmitters and neuro-developmental action. It is thought that levetiracetam may change the volume of discharge of neurotransmitters from the pre-synaptic cell to the synaptic cleft through its action on SV2A. Neurotransmitters, such as dopamine and noradrenalin are known to be neuroprotective if the quantity of antipsychotic agents is a minimal,36 although neurodeficiency will occur if it is excessive.37 In epileptic patients, higher BDNF and Bcl-2 levels provide a neuroprotective effect similar to that seen in childhood, and therefore gives a wider range to evaluate the input stimuli as appropriate. Levels of neurotransmitters released from the pre-synaptic neuron are changed by levetiracetam-treatment and cause post-synaptic neurons to undergo neuro-maturation. This pruning of the aberrant excitable network exerts the anti-epileptogenesis effect of levetiracetam treatment (Figure 4D). We therefore suggest that the mechanism of acquired anti-epileptogenesis by levetiracetam treatment is the same as the spontaneous recovery seen in idiopathic generalized epilepsy during adolescence. The current findings verify our assertions, although the process of GSK-3β activation following the volumetric alterations of neurotransmitter release within the synaptic cleft is not well established. Therefore further research is needed to determine which neurotransmitter and which receptor are involved in the process (Figure 3).
Acknowledgments
We are grateful to Dr. T. Nakagawa for his helpful advice and to UCB S. A. levetiracetam supply.