INTRODUCTION
Autism spectrum disorders (ASD) is characterized by impairments in reciprocal social interaction, communication and by restricted/repetitive behaviors in early childhood.12 ASD affects about 1 in 80–110 individuals, with onset before the age of three years.134 In addition to specific clinical symptoms, approximately 31% ASD patients' also present intellectual disability (ID) and 20–25% has seizures.567 There is no definitive pharmacotherapy for the treatment of core symptoms of ASD. Therapies commonly used to reduce behavioral, educational and immunological symptoms. Antipsychotic drugs only target the secondary symptoms of irritability, aggression, anxiety, depression, and self-injurious behaviors.28 Until now, family-based and twin studies indicated significant genetic basis for ASD susceptibility.13 Etiopathogenesis of ASD remains largely unknown but now, it is well recognized that ASD as a complex disorder, involve the complex interaction of several genes and environmental risk factors.91011 The genetic architecture of ASD comprises a diversity of rare single nucleotide variants, copy number variations (CNVs), chromosomal abnormalities, and common polymorphic variations.12 The recent advances in ASD genetics are summarized in Table 1. Additionally epigenetic dysregulation might contribute to significant proportion of ASD cases.21113
Advances in genomic technology, including array-based genome-wide association studies (GWAS) of single nucleotide polymorphisms (SNPs), CNV studies and whole exome-sequencing (WES) technologies, have been key to this paradigm shift in psychiatric research.12141516 Like other complex diseases such as hypertension and diabetes, psychiatric disorders are not inherited in a Mendelian fashion but have a complex genetic architecture involving a spectrum of mutations from small DNA sequence variations to large chromosomal rearrangements and epigenetic regulation.1718 According to latest data, rare truncating heterozygous variants have a predominant role in the etiology of autism.19 This review briefly emphasizes autism genetics, examining recent evidences by demonstrating the potential usage of next generation sequencing technologies, particularly WES studies in this respect.
DEVELOPMENT OF NEW SEQUENCING TECHNOLOGIES
In the 1970s, Sanger et al.20 and Maxam and Gilbert21 developed new methods to sequence DNA by chain termination and fragmentation techniques, respectively. The technique developed by Sanger and colleagues, commonly referred as Sanger sequencing, required less handling of toxic chemicals and radioisotopes than Maxam and Gilbert's method. As a result, it became the prevailing DNA sequencing method for the next 30 years.2223 Even union of chain termination sequencing by Sanger et al. and the polymerase chain reaction (PCR) by Mullis et al. established many marked events in genetic studies, eventually the completion of Human Genome Project, as well as other genome projects of many species.22242526
There is a great diversity of DNA variation in human genome and these variations comprise small base changes (substitutions), rearrangements such as inversions and translocations, insertions and deletions of DNA and large genomic deletions of exons or whole genes. Traditional sequencing methods, especially Sanger sequencing, is restricted to the discovery of variations including small insertions or deletions. For the other remaining mutations dedicated assays are frequently performed with fluorescence in situ hybridization (FISH) for conventional karyotyping, or comparative genomic hybridisation (CGH) microarrays to detect submicroscopic chromosomal CNVs such as microdeletions or duplications.27
Developments between 1977 and 2005 were focused on increasing through as, accuracy and decreasing cost. Automated Sanger sequencing method defined as the first-generation technology, while Next Generation Sequencing (NGS) represents the second generation technology. The need for high-throughput, low-cost sequencing drove the development of massively parallel technologies. As a result, new improvements for sequencing technologies developed in recent years.28 NGS libraries in a cell free system were the first major difference. Thousands-to-many-millions of sequencing reactions are produced in parallel which is called "massively parallel sequencing."29 Also, the sequencing output is directly detected without the need for electrophoresis; base interrogation is performed cyclically and in parallel.2225 The huge number of reads generated by NGS enabled the sequencing of whole genomes with an unprecedented speed and thus it became widely used in various fields of life sciences.25 The first NGS technology to be released in 2005 was the pyrosequencing method by Roche 454 Life Sciences. There are other sequencing platforms including Illumina, Solex, SOLID.22303132 Third-generation sequencing technology can determine the base composition of single DNA molecules and also enables real-time sequencing.25
NGS involves the whole of genome and whole exome sequencing, aim at developing an understanding the variations in human genome, determines the genetic tendencies to diseases and finding out the pharmacogenetical drug responses. With the evolution of NGS technology, costs of all the genome sequencing activities will gradually decrease. NGS includes different applications including RNA sequencing, ChIP-seq, ChIP-chip, whole genome sequencing, whole genome structural variations, mutation detection and carrier screening, determination of hereditary diseases, preparation of DNA libraries, mitochondrial genome sequencing and individual genomics.33 This new generation DNA sequencing technology has provided certain benefits in terms of acquiring information pertaining to the genetic/epigenetic regulatory networks, chromatin structure, nuclear structuring and genome variations.2234 Undoubtedly, these kinds of research activities will be at the forefront in the future years.
NGS like many other techniques has some limitations. Exome enrichment is the basis for exome sequencing technology. The exome regions are enriched by different methods including hybridization capture or solution based methodology.35 Differently from Sanger sequencing, each run of samples generated a single sequencing read. The specific location of each read must be determined computationally referred as mapping or alignment. Secondly, multiple coverage is required to analyze the full allelic content of the sample.29 Fragment sequencing has higher sequencing error rates than Sanger sequencing; thus, further validation of identified variations using Sanger sequencing is very important.36 The weaknesses of these technologies are solved with the arrival of third-generation sequencing technologies.35 HapMap or dbSNP databases, cannot effectively exclude common and irrelative variants that obtained from WES. With the application of the Bonferroni correction and true bioinformatic approaches, the true variants would be distinguished through the yielded threshold.37 Statistical metrics are essential to distinguish false positive data from true positives.38 The researchers should be focused on the nonsynonymous (NS) variants, splice acceptor-site or donor-site mutations, and frameshift mutations (insertions/deletions).
Whole exome sequencing in Mendelian disorders and complex diseases
Determination of the genetic basis which lies behind the rare single gene diseases is important for understanding to the mechanism of disease in terms of finding out its role in the biological pathways and developing the treatment.37 Since researches on wide genome linkage and Mendelian inheritance pattern (autosomal dominant, autosomal recessive, X-linked recessive) while they demonstrate a perfect segregation, because of this, successful results were obtained in the determination of these variants.3940 Exome sequencing serves for the sequencing of all the exons in the genome. Since a significant part of mutations exists in the exons, this approach is efficient in revealing the Mendelian diseases. Success of exome sequencing in revealing these mutations and identifying genes have been demonstrated by several studies.404142 It is estimated that 85% of the disease-causing mutations are located in coding and functional regions of the genome.43 Therefore, sequencing of the exome, has the potential to uncover the causes of large number of rare, mostly monogenic (Mendelian disorders), genetic disorders as well as predisposing variations in common diseases.24
Within the last five years, researches have made numerous studies regarding the complex diseases. Before these researches, complex phenotype studies mainly focused on the candidate gene, linkage and association studies. Although the linkage analysis were used to define the variants in thousands of Mendelian diseases, this method was not effective in complex diseases due to the effect of many genetic and environmental factors. Additionally, exome sequencing provides new opportunities in complex and rare diseases, and also in sporadic cases, which have been used since 2007.44 However, currently only the allelic variants responsible from only less than half of these diseases have been determined. Factors aggravating the diagnosis include the existence of only one case within the family and locus heterogeneity (mutations in different locus result in the same phenotype). Each of these factors limit the strategies used in candidate gene determination.3940 Due to the difficulty in finding sufficient number of samples, linkage studies are not effective in focusing on the rare diseases and sporadic cases. In addition, linkage studies may also fail due to the genetic and phenotypic heterogeneity in Mendelian diseases. Exome sequencing has been used in defining the variants of several rare diseases such as the Kabuki Syndrome, Miller Syndrome and the Schinzel-Giedion Syndrome. WES may be considered as an efficient technique in diseases which demonstrate genetic heterogeneity.3842 GWAS of neuropsychiatric disorders have unequivocally shown that common variants with large effects do not underlie schizophrenia or autism.45 For this reason, it is important to use such technologies to discover new molecular pathways which are responsible for these complex disorders.
Whole exome sequencing implications in autism
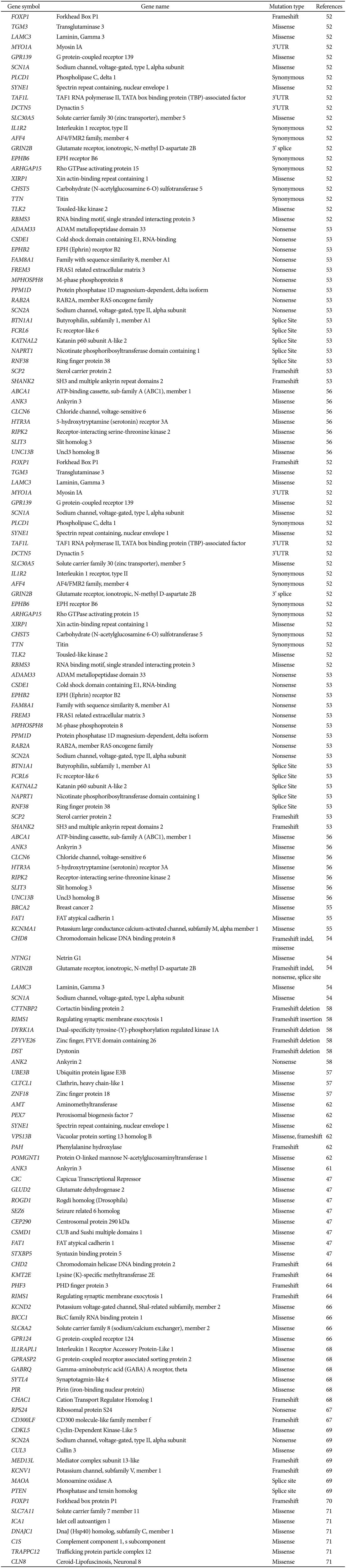
With the advances in genomic technology, such as array-based GWAS and array comparative genomic hybridization (aCGH), researchers have been able to detect several rare genomic rearrangements and CNVs in subsets of cases with ASD.18 GWAS and WES have allowed the potential genetic mechanisms underlying the overlap between psychiatric disorders to be investigated more directly and easily.1846 WES seems to be frequently used in complex disorders including autism and the number of such studies is increasing gradually (Table 2).475051 One of the advantages of WES is that a specific gene which harbors a genetic variant can be identified. Additionally, related biological pathways can be further investigated. The function of specific types of mutation, including nonsense and missense mutations can be predicted by the help of publicly available bioinformatic resources.14
The exomes of 20 individuals with sporadic ASD patients and their parents (trio-based study) were sequenced. Totally, 21 de novo mutations were identified and 11 of these mutations were altering the protein structure. The researchers suggested that trio-based exome sequencing is a powerful approach for identifying new candidate genes for ASD.52 Sanders et al.53 showed a total of 279 identified de novo coding mutations using WES of 928 individuals. Interestingly, two independent nonsense variants disrupt the same SCN2A gene. A total of 677 individual exomes from 209 families were sequenced in 2012. Moreover, 39% (49 of 126) of the most severe de novo mutations were related with a highly interconnected β-catenin/chromatin remodelling protein network as new candidate genes for autism. In probands' exomes, protein-altering mutations were observed in CHD8 and NTNG1 genes.54 In another study, Neale et al.55 assessed the role of de novo mutations by sequencing the exomes of ASD cases and their parents (n=175 trios). They observed totally 161 coding region point mutations (50 silent, 101 missense, and 10 nonsense), 2 conserved splice site (CSS) single nucleotide variations (SNVs) and 6 frameshift indels. Their results provided strong evidence in favor of CHD8 and KATNAL2 genes were likely to be important genetic risk factors. In the same year another WES study was conducted in a cohort of 20 ASD patients. They also sequenced an additional 47 ASD samples, and identified three different missense mutations in ANK3 gene in four unrelated ASD cases. One of the mutation (c.4705T>G/p.S1569A) is a de novo mutation. With this finding the authors suggested an association between ANK3 mutations and ASD susceptibility and imply a shared molecular pathophysiology between ASD and other neuropsychiatric disorders such as schizophrenia, and bipolar disorders. ANK3 gene is a member of the ankyrin family of proteins that is associated with the spectrin-actin cytoskeleton in neuronal cells. Loss of function of ANK3 may influence neuronal excitability through ion channel function and affects synaptic development and functions.56
WES of 16 probands by the completion of homozygosity mapping revealed validated homozygous, potentially pathogenic recessive mutations that segregated perfectly with disease in 4 families. Identified mutations except in the NCKAP5L gene (the other candidate genes were UBE3B, CLTCL1 and ZNF18) were probably or possibly damaging according to PolyPhen 2. The candidate genes that identified in this study (UBE3B, CLTCL1, NCKAP5L, ZNF18) encode proteins involved in different pathways especially proteolysis, GTPase-mediated signaling, cytoskeletal organization.57 Exome sequencing of simplex 343 families with ASD revealed de novo small indels and point mutations. This study demonstrated that gene-disrupting mutations (nonsense, splice site, and frame shift) are twice as frequent in affected versus unaffected children.58 In another study a large autism family with five generation (47 family members) were investigated with both WES and linkage analysis. The authors obtained strong association for localization of a risk locus to chromosome 22, precisely bound the interval likely to carry the risk variant, and prioritize evaluation of all exome sequence variants within that region.59 Exome sequencing was applied to the X chromosome in 12 unrelated families with two affected males with a different approach. A nonsense mutation in the TMLHE gene was identified in two brothers with autism and intellectual disability. Further functional analyses confirmed that the mutations were associated with a loss-of-function and this finding supported the rare variants on the X chromosome are involved in the etiology of ASD.60 With WES, two probands from a large pedigree including two parents and eight children examined. The researchers identified 59 candidate variants that may increase susceptibility to autism. They suggested only one gene, ANK3 (c.11068G>A/p.G3690R mutation), as the most likely candidate gene in this family.61 WES applied to 11 ASD families enriched for inherited causes due to consanguinity and this study determined familial ASD associated with biallelic mutations in disease genes (PAH, SYNE1, AMT, POMGNT1, PEX7, VPS13B), some of them implicated for the first time in ASD. PEX7 gene encodes a receptor required for import of PTS2 (peroxisome targeting signal 2)-containing proteins into the peroxisome.62
Total of 42 Australian ASD families with 48 probands were screened with WES. Among 44 de novo variants, there were 4 were intergenic, 29 associated with protein-coding sequences, 9 were intronic, and 2 occur in 3'-UTR regions of genes. Gene ontology analysis revealed that identified de novo variants cluster in key neurobiological processes involving neuronal development, signal transduction and synapse development including the neurexin trans-synaptic complex.63 Cukier et al.48 performed WES on 164 individuals from 40 families with multiple affected individuals. According to their data, potentially novel ASD candidates identified that including genes with previous clinical and molecular evidence supporting a neuronal function such as RODG1, CIC, NTSR2, GLUD2 and SEZ6. Additional variations were also found in previously reported autism candidate genes including CEP290, CSMD1, FAT1, and STXBP5. WES data from 787 ASD families (2,963 individuals), two ASD-associated genes, KMT2E (MLL5), a chromatin regulator, and RIMS1, a regulator of synaptic vesicle release were identified in this study.64 WES was performed with 488 ASD cases and 372 controls of European ancestry in 2014. The authors identified variant in one gene, Fanconi-associated nuclease 1 (FAN1) has being associated with schizophrenia and ASD. They suggested that FAN1 is a key driver in the 15q13.3 locus, encoding a DNA repair enzyme, for the neurodevelopmental and associated psychiatric phenotypes.65 In the same year another study of WES was published on a family with identical twins affected with autism and intractable seizures. A de novo variant was identified in the KCND2 gene which encodes the Kv4.2 potassium channel and mutant protein disrupts potassium current inactivation. This alteration (p.Val404Met) is novel and occurs in a highly conserved region.66 In a recent study where two rare heterozygous truncating variations, Q191X in RPS24 and P261fsX266 in CD300LF have been identified as risk candidates for ASD. RPS24 gene encodes ribosomal protein 24 (RPS24), which is involved in ribosome biogenesis. CD300LF is a member of the CD300 gene complex and encodes CMRF35-like molecule 1 (CLM-1), which is an immunoreceptor expressed on myeloid cells. CLM-1 overexpression reduced acute brain injury therefore CLM-1 has protective effects against neuroinflammation.67 Thirthy females with autism from multiplex families have been selected to WES case study. Functional variants of X-linked genes (GABRQ, IL1RAPL1, PIR, GPRASP2 and SYTL4) were identified in 5 females.68 The latest study with exome sequencing was conducted with 36 males with a diagnosis of idiopathic ASD. Five de novo (SCN2A, MED13L, KCNV1, CUL3, and PTEN) and two inherited X-linked variants (MAOA and CDKL5) were identified in seven cases. CDKL5 gene is a member of the Ser/Thr protein kinase family and encodes a phosphorylated protein with protein kinase activity. PTEN gene is a part of the PI3K-AKt and PDGF signaling pathways. SCN2A gene is involved in axon guidance and L1CAM interaction pathways.69 A heterozygous de novo FOXP1 variant (c.1267_1268delGT, p.V423Hfs*37) was identifed with exome sequencing in a patient with autism, intellectual disability and severe speech and language impairment. This variant disrupts FOXP1 activity, including subcellular localization and transcriptional repression properties.70 Egawa et al.71 performed WES in two families with three affected siblings. Six novel missense variants were determined with this study including SLC7A11, ICA1, DNAJC1, C1S, TRAPPC12 and CLN8 genes. They suggested that CLN8 is a potential genetic risk factor for ASD and this gene plays a role in cell proliferation during neuronal differentiation and in protection against neuronal cell apoptosis.
CONCLUSION
The arrival of NGS and WES methods comprise the beginning of a new era, not just for autism research but also for researches into nearly every complex disorder. With new sequencing data, scientists will benefit greatly from the ability to combine genetic data generated by multiple methods (for example, combining GWAS and/or linkage data with WES data). WGS will become increasingly more cost-effective as sequencing costs decrease and bioinformatics tools improve, and these advances will open up the possibility of detecting non-coding genetic changes in regulatory regions in the genome.14 The WES approach has some limitations as it only detects individual genetic variations based on single nucleotide polymorphisms (SNPs) and small indels associated with exonic regions of the genome. WES technology does not detect large CNV or genetic variants of regulatory sequences located in intergenic regions. Compared to traditional methods, exome sequencing is a less costly and a faster technique for the determination of mutations as well as for autism and in other neurological diseases where lots of candidate genes and loci are responsible from these disorders. WES allows scanning the mutations/variations in genes commonly observed in many diseases with genetic heterogeneity.72 As a result, it is an undeniable fact that WES will also be used as a golden standard for more complex diseases as well as ASD and be used with all genome sequencing activities, along with their use for revealing the underlying genes of the rare genetic disorders in the future.