The Association of Blood-Based Inflammatory Factors IL-1β, TGF-β and CRP with Cognitive Function in Alzheimer’s Disease and Mild Cognitive Impairment
Article information

Abstract
Objective
Many patients suffer from dementia in its most common form, Alzheimer’s disease (AD). In this study, the levels of IL-1β, TGF-β and CRP, which are involved in the inflammatory response in Alzheimer’s disease and its mild cognitive impairment (MCI), were measured and analyzed.
Methods
Seventy nine subjects participated in this study (mean age: 75.56 years, female: 54.3%, AD: 26, MCI: 28, normal: 25). The overall cognitive function of the subjects and the severity of the disease stage were assessed using the Mini-Mental State Examination (MMSE-K), the Clinical Dementia Rating (CDR), the Global Deterioration Scale (GDS) and the Geriatric Depression Scale-Korean (GDS-K).
Results
It was observed that patients with AD had significantly higher levels of IL-1β and TGF-β than the patients with MCI and normal controls. In addition, the MCI group showed a statistically significantly higher TGF-β concentration than the normal group.
Conclusion
These results suggest that IL-1β and TGF-β may be useful biological markers for patients with Alzheimer’s disease.
INTRODUCTION
Many patients suffer from dementia in its most common form, Alzheimer’s disease (AD), which is characterized by general cognitive impairments in cognitive domains such as memory, time-space orientation, working memory and language. AD increases the suffering and burden of patients and caregivers because of the behavioral and psychological symptoms of dementia (BPSD) and impairment in the activity of practical living [1].
The number of AD patients is expected to increase to more than 90 million worldwide by the year 2050, with the recent rapid increase in people’s life expectancy [2]. Although not yet clear and consistent, some risk factors have been identified; Age is known to be one of a major risk factor for AD. Epidemiological studies over the past several decades have found AD risk factors such as depression, hypothyroidism, drinking, and vitamin deficiencies [3]. Until recent years, the diagnosis of AD was based on clinical assessment, medical history, physical examination, psychological assessment, blood tests, and radiological examinations [4]. However, the way we diagnose AD is changing, with a recent and particular focus on the pathological neurodegenerative outcomes that are known before clinical manifestations [5]. Furthermore, to effectively manage the neurodegenerative disease, identifying a biomarker will be of great help in diagnosis of the disease, and will contribute greatly to treatment.
The two main neuropathologies of AD are extracellular betaamyloid (A beta) and intracellular abnormally phosphorylated tau [6]. The production of Aβ is a result of cleavage of amyloid precursor protein (APP) as a significant pathologic finding, and the high level of Aβ is in AD [7].
Tau is a component of the microtubule, which produces hyperphosphorylated bundles of nerve fibers in AD [8]. Until recently, interventions and treatments targeting such neurodegenerative chain reactions have not been successful. Drug therapy for this has only a slight effect and does not show satisfactory improvement in symptoms [9]. Therefore, there is a need to develop alternative or ancillary treatment strategies based on other etiologic hypotheses, with the potential to modulate disease. Currently, there are a number of reasons why inflammatory reactions can be involved in neurodegenerative cascade in AD [10]. Inflammation is a reaction that eliminates both the necrotic cells and tissues resulting from the initial injury as well as the causes of cell damage. If the cells or tissues are not recovered, the inflammation will become chronic and continue to damage surrounding tissues, causing severe tissue deterioration [11].
Brain inflammation is a hallmark of AD, but inflammatory symptoms such as fever, pain, and swelling do not appear in the brain [12]. In addition, in AD, the brain usually exhibits chronic inflammatory conditions instead of acute inflammation. Tissue with chronic inflammation characterizes an increased number of inflammatory cytokines, including AD [13].
IL-1 acts as an initiator of the immune response and plays an important role in complex hormone and inflammatory chain reactions [14]. There is also evidence that IL-1 is associated with nerve damage in both acute neurodegenerative diseases such as stroke and chronic neurodegenerative diseases such as AD [15].
In particular, IL-1β has direct neurotoxicity among IL-1 subtypes and it is also known to stimulate the production of Aβ 42 and Aβ 40 by affecting the metabolism of beta-amyloid precursor protein (βAPP) [16]. These IL-1β have also been found in higher concentrations in peripheral blood tests of AD patients than in the normal group [17]. However, the peripheral blood concentration of IL-1β was reported to be not different from that of the normal group, so further studies are needed [18].
TGF-β is an inactivated precursor, a cytokine that needs activation to exert its effects and is an important regulator of cell proliferation, differentiation, and formation of extracellular matrix [19]. TGF-β inhibits the proliferation and differentiation of immune cells and plays an important role in the growth and survival of neurons in the brain of an AD sufferer [20]. TGF-β, on the other hand, has been shown to increase β-amyloid in rat models and is thought to exacerbate the pathogenesis of amyloid formation [21]. TGF-β has also been shown to be elevated in the central nervous system of AD patients [22]. TGF-β is known to regulate a wide variety of processes such as brain injury response, brain inflammatory response, extracellular matrix production, distribution of amyloids and inhibition of cell death [23].
CRP is representative of acute phase reactions that respond nonspecifically to inflammation or tissue damage [24]. The presence of acute-phase reactive substances, such as CRP, has been identified in posttraumatic brain studies in AD patients [25]. The relationship between AD and inflammation is unclear, but there was a study tried to investigate the association between AD and CRP [26].
Efforts to identify inflammatory biomarkers are still ongoing. The concept and efficacy of biomarkers make diagnosis and treatment of disease easier. There were many studies to identify the biomarkers of AD through cerebrospinal fluid (CSF), and the results have been published [27,28]. However, since CSF should be obtained by spinal tapping, it is difficult to collect and test specimens. And there is a limit to doing so [29].
This study tried to compare IL-1β, TGF-β and CRP levels in patients with AD, those with mild cognitive impairment (MCI) and a normal group. In each of these three groups, cognitive function was assessed by the Mini-Mental State Examination (MMSE-K), Clinical Dementia Rating (CDR), the Global Deterioration Scale (GDS) and the Geriatric Depression Scale. This study aimed to investigate the relationship between the results of scales and blood-based inflammatory factors. Since there have been few studies on blood-based inflammatory factors in dementia so far, the results of this study are expected to be of great help in the pathology, evaluation, prognosis, and treatment of AD and MCI.
METHODS
Research subjects
This study was carried out from May 2017 to March 2018 based on the clinical judgements of psychiatrists, neurocognitive function evaluations, and radiological examinations such as MRIs among those who visited the Department of Psychiatric Medicine and Dementia Clinic of Inje University Ilsan Paik Hospital. Research subjects of this study include both inpatients and outpatients who visited the clinic of Ilsan Paik Hospital. Twenty six patients with diagnosed AD, 28 patients with MCI, and 25 normal subjects were analyzed for blood inflammatory factors and data. The sample size of this study is G* Power 3.1. As a result of analysis of the effect size γ=0.40, α=0.05 and power (1-β)=0.8 using the program, the appropriate sample number was 66 [30]. In this study, 79 patients were selected for consideration with a 20% dropout rate [31].
This study was reviewed and approved by the Medical College of Inje University Ilsan Paik Hospital Clinical Research Ethics Committee (ISPAIK 2017-03-008). The study received consent for the information content from the study subjects.
Subjects selection criteria
This study was conducted from May 2017 to March 2018 for those who visited the Department of Psychiatric Medicine and Dementia Clinic of Inje University Ilsan Paik Hospital. Based on the clinical judgements of psychiatrist, psychiatric evaluation, and radiological findings such as those obtained through MRI, we classified AD, MCI and normal control group.
AD was diagnosed according to the criteria of the National Institute of Neurological and Communicative Disease and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA). For the diagnosis, history taking, physical examination and cognitive function tests were performed. Sex, age, and physical illness of the subjects were also examined.
MCI was classified according to the diagnostic criteria proposed by Petersen et al. [32] The MCI group included subjects who had a subjective complaint of memory impairment and whose memory was less than that of person the same age or with the same education level but did not fit the criteria for dementia because the overall cognitive function was maintained and their daily activities were not interrupted. In this study, healthy control subjects were participants who were living in a community with no cognitive decline. The overall cognitive function of the subjects and severity of their disease were assessed by MMSE-K, CDR, GDS, and Geriatric Depression Scale-K (GDS-K).
Subjects exclusion criteria
Exclusion criteria for this study were: 1) diagnosed with dementia due to causes other than Alzheimer’s disease, 2) diagnosed with Alzheimer’s disease accompanied by other dementia, such as vascular dementia or frontotemporal dementia, 3) history of head trauma or brain damage, 4) other neurodegenerative diseases such as Parkinson’s disease or Huntington’s disease, 5) history of substance abuse, 6) medical problem that may impair cognitive function, such as thyroid dysfunction, hematologic disease, or renal disease, 7) accompanying psychotic or mood disorder.
Methods
Blood sampling were taken in the morning after overnight fasting. Blood tests were conducted at Seoul Clinical Laboratories. Inflammatory factors such as IL-1β, TGF-β, and CRP were examined.
Evaluation scales
Mini Mental State Examination-Korean (MMSE-K) is a Korean version of MMSE that uses standardized questions to measure cognitive function. MMSE-K consists of 7 categorizations including time and spatial orientation, memory registration, memory recall, attention and calculation, language function, understanding and judgment, and total 30 items [33].
CDR is a tool for measuring cognitive and social functions of patients through interviews with patients and caregivers. CDR includes items such as memory, orientation, judgment and problem-solving, community affairs, home and hobbies, and personal care. These six sub-items are evaluated on five levels of 0, 0.5, 1, 2, and 3, respectively, and the overall score is determined [34].
The GDS is an evaluation tool used for measuring behavioral abnormality and daily life as well as cognitive function. According to the GDS, the degree of cognitive decline increases progressively up to stage 7 [35].
The Geriatric Depression Scale was developed in 1983 as a tool for evaluating depression and it is specialized for the elderly population. It is an evaluation item considering delays in the psychomotor speed of the elderly depression and performance of cognitive function tests [36]. In Korea, the Geriatric Depression Scale of 30 items was translated into the GDS-K in 2004 [37].
Statistics
All subjects were divided into three groups according to their level of cognitive function: AD, MCI and normal, and analyzed according to gender and age. Differences according to the gender of each group were evaluated using the chi-square test. One-way ANOVA was used to analyze age differences among the three groups. One-way ANOVA was also used in MMSE-K and GDS-K to confirm whether there was a significant difference between the three groups. In overall CDR and the GDS, the chi-square test was used to determine whether there was a significant difference in the three-group scale scores. The differences in IL-1β, TGF-β, and CRP concentrations in each group were analyzed.
The Kruskal-Wallis test was performed for IL-1β and CRP because they did not satisfy the normality conditions. The Mann-Whitney test was used for post-hoc analysis when significant differences were observed between the three groups. TGF-β met the normal distribution and one-way ANOVA was performed to identify significant differences in TGF-β between the three groups. When there was a difference between the three groups, Tukey Honestly Significant Difference (HSD) was performed as post-hoc analysis.
To determine whether IL-1β, TGF-β, and CRP levels correlated with cognitive function scale scores, MMSE-K, GDS-K and Pearson correlation analysis was performed. In addition, correlation analysis was performed using Spearman’s rho to determine whether the levels of IL-1β, TGF-β, and CRP correlated with overall CDR and the GDS. The collected data were analyzed using Statistical Package for the Social Sciences (SPSS) ver. 20.0 (IBM Corp., Armonk, NY, USA). P<0.05 was the level used to determine statistical significance.
RESULTS
Demographic and clinical data
Among the 79 subjects, 26 subjects were diagnosed AD, 28 subjects MCI, and 25 subjects were normal. There were 43 (54.3%) female and 36 male (45.7%) subjects, and the gender difference in the groups was not statistically significant (p=0.164). The mean age was 75.57±6.12 years. The mean age of the groups was 75.54±6.17 years in the AD group, 75.60±6.15 years in the MCI group, and 75.56±6.29 years in the normal group. There was no statistically significant difference among the groups (p=0.999). The mean MMSE-K in each group was 15.85±5.04 in the AD group, 22.79±2.91 in the MCI group, and 26.68±1.95 in the normal group, and the mean total MMSE-K was 21.73±5.66. There was a statistically significant difference in MMSE-K scores among the three groups (p<0.001). The mean GDS-K score of each group was 13.42±7.62 for the AD group, 10.18±6.48 for the MCI group and 10.20±7.49 for the normal group. The mean total GDS-K score of the three groups was 11.25±7.26. There was no statistically significant difference among the three groups. These results are shown in Table 1.

Demographic and clinical data of subjects
The mean CDR of each group was 1.13±0.59 for the AD group, 0.50±0.19 for the MCI group and 0.40±0.20 for the normal group. The mean overall CDR of all three groups was 0.73±0.69. The overall CDR showed statistically significant differences among the three groups. The result of overall CDR is shown in Table 2.
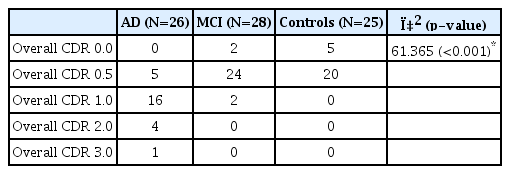
Clinical overall CDR data of subjects
The mean levels of IL-1β in each group were 4.74±2.10 for the AD group, 2.06±2.38 for the MCI group and 0.89±1.61 for the normal group. In the Mann-Whitney post-hoc analysis after the Kruskal-Wallis test for IL-1β, the AD group was found to have statistically significantly higher levels than the MCI group (p<0.001) and the normal group (p<0.001). There was no statistically significant difference between the MCI group and the normal group (p=0.019).
As regards TGF-β levels, these were 56.29±9.84 in the AD group, 42.74±11.57 in the MCI group and 36.13±7.44 in the normal group. In the Tukey HSD post-hoc analysis for TGF-β, the AD group’s TGF-β level was found to be statistically significantly higher than that of the MCI group (p<0.001) and the normal group (p<0.001). In addition, the level of TGF-β was significantly higher in the MCI group (p=0.044) than in the normal group.
The mean CRP level in each group was 0.26±0.45 for those with AD, 0.16±0.14 for those with MCI and 0.12±0.06 for the normal group. There was no statistically significant difference among the groups according to the Kruskal-Wallis test for CRP. The level of inflammatory factors in subjects are shown in Table 3.

Inflammatory factors of subjects
Correlation between IL-1β, TGF-β, CRP levels and cognitive function
MMSE-K scores were highest in the normal group, followed by the MCI group and lowest in the AD group. The MMSE-K scores were significantly different among the groups (p<0.001).
There was a significant negative correlation between IL-1β and MMSE-K scores in all groups (r=-0.454, p<0.001) in Figure 1. There was a significant positive correlation between IL-1β and overall CDR (rho=0.452, p<0.001). IL-1β and the GDS scores also showed a significant positive correlation (rho=0.352, p=0.001). There was also a positive correlation between IL-1β and GDS-K scores although it was not significant (r=0.119, p=0.296).

Correlation between IL-1β levels and MMSE in all subjects (r=-0.454, p<0.001). Significant at p<0.05. MMSE-K: Mini Mental State Examination-Korean.
TGF-β and MMSE-K scores showed a significant negative correlation (r=-0.395, p<0.001) in Figure 2.
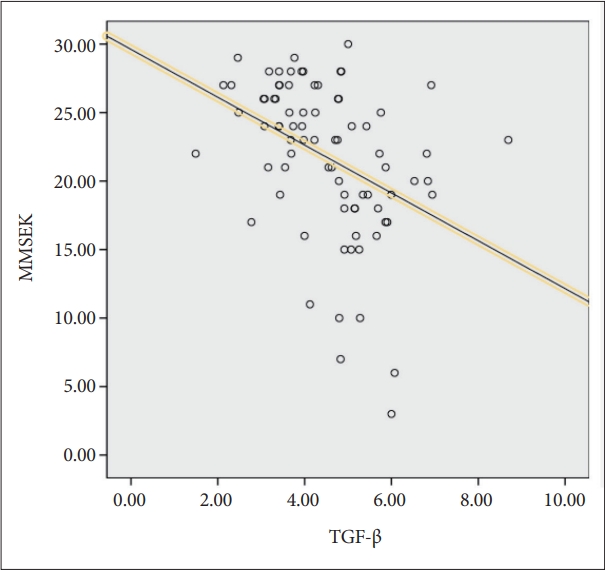
Correlation between TGF-β levels and MMSE in all subjects (r=-0.395, p<0.001). Significant at p<0.05. MMSE-K: Mini Mental State Examination-Korean.
Conversely, there was a significant positive correlation between TGF-β and overall CDR (rho=0.418, p<0.001). TGF-β and GDS scores also showed a significant positive correlation (rho=0.357, p<0.001). On the other hand, there was no significant correlation between TGF-β and GDS-K scores (r=0.178, p=0.116).
There was no significant correlation between CRP and MMSE-K scores in the whole group (r=-0.136, p=0.232). CRP and overall CDR were also not significant (rho=0.133, p=0.242). CRP and GDS scores had no significant correlation (rho=0.171, p=0.131). Correlation between CRP and GDS-K scores was not significant (r=0.211, p=0.062).
DISCUSSION
Several studies have reported that inflammation affects the pathology of AD. Pro-inflammatory cytokines alter the expression and processing of the β-amyloid precursor protein [38,39].
Licastro et al. [40] found that IL-1β levels in patients with AD were significantly higher than in normal groups. Reale et al. [41] also suggested that IL-1β was significantly increased in unstimulated peripheral blood mononuclear cells of patients with AD compared with normal controls.
Chao et al. [42] reported that patients with post-mortem AD had twice the TGF-β concentration in CSF compared with normal controls in age-sex matched groups (p<0.05), and peripheral blood TGF-β levels were significantly higher than those of the normal group (p<0.001). Malaguarnera et al. [43] reported that TGF-β levels were significantly elevated in patients with AD and vascular dementia compared with normal controls.
However, Mocali et al. [44] reported that TGF-β was reduced in AD patients and discusssed that reduced TGF-β in AD patients can potentiate pro-inflammatory cytokines that are activated or expressed by microglia and astrocyte.
Mancinella et al. [45] found that inflammation plays an important role in dementia, and found that patients with dementia had higher CRP levels than normal controls.
Several studies have shown that patients with AD exhibit higher levels of inflammatory factors. The results of this study suggest that IL-1β and TGF-β levels were higher in patients with AD. Therefore, measurement of IL-1β and TGF-β, which are inflammation-related factors, may help with early detection and early treatment of dementia. Appropriate management and treatment of inflammation are thought to help prevent dementia.
In this study, we examined the relationship between inflammatory factors such as IL-1β, TGF-β, and CRP and cognitive functions such as MMSE-K, overall CDR and the GDS. The higher the levels of IL-1β, TGF-β, and CRP, the lower the cognitive function. The results of this study are consistent with thefinding that the pathology of cognitive impairment is caused by inflammatory mechanisms in patients with cerebrovascular disease [46]. In addition, due to the fact that brain inflammation is an important factor in neurodegenerative disorders in many studies, the level of inflammatory factor may be high, while the cognitive function is low [47].
The pathogenetic mechanism of IL-1β, TGF-β, and CRP in AD remains unclear. However, there are some hypotheses that support this study. Pro-inflammatory cytokines such as IL-1β may be related to neurodegenerative cascade of AD. Microglia, the resident immune cells of the brain, constantly surveys the microenvironment under psychological conditions [48]. In AD, deposition of amyloid-β plaques initiates a spectrum of cerebral neuroinflammation mediated by activating microglia. And then activated microglia may play an important role in the secretion of pro-inflammatory cytokines such as IL-1β, IL-6 [49]. These cytokines released from brain cells, also stimulates the biosynthesis of amyloid-β plaques [50]. As a result, IL-1β may be increased in Alzheimer’s disease patients.
Another possible mechanism associated with peripheral pro-inflammatory cytokine such as IL-1β is their ability to induce the production of indoleamine 2, 3-dioxygenase (IDO) [51]. IDO catalyzes the rate-limiting step in the synthesis of kynurenine from tryptophan, and kynurenine synthesis in peripheral blood significantly affects kynurenine concentration in the central nervous system (CNS) [52]. The increase in quinolinic acid, the neurotoxic metabolite of kynurenine, was higher in patients with AD than in the normal group [53]. The pathological significance of quinolinic acid in patients with AD was suggested due to its ability to induce hyperphosphorylation of tau [54]. The activation of IDO was significantly increased in AD patients, and the degree of IDO activity was significantly correlated with peripheral inflammatory factors and cognitive decline [55].
The role of anti-inflammatory cytokine such as TGF-β is unclear in AD. Activated microglia play a key neuropathogenic value in AD. And this anti-inflammatory cytokine may exert a neuroprotective effect by inhibiting the inflammatory response of microglia [56]. TGF-β also may have a neuroprotective effect by stimulating the synthesis and release of nerve growth factor [57]. TGF-β binds β-amyloid precursor protein, which could reduce the availability of the neurotoxic constituent β-amyloid [58]. In conclusion, elevated TGF-β levels in AD may represent a protective host response to inflammatory injury of neuron.
Previous studies on the association of inflammatory factors with cognitive function have been performed mostly on patients with dementia. However, this study expanded the scope of study by including the MCI group. The IL-1β and TGF-β concentrations in the MCI group were significantly lower than those in the AD group. In particular, IL-1β levels were not significantly different between the MCI group and the normal group but TGF-β levels were significantly higher in the MCI group than in the normal group.
In this study, we can think that TGF-β can be one indicator of differences in normal, MCI, and AD groups. It is also thought that the study of TGF-β could be the future direction of treatment in AD. Indeed, studies are being done in this regard. Research is being conducted on blocking the TGF-β in pathways in peripheral macrophages can signally clear up β-amyloid plaque in Brain [59].
There are some limitations to this study. First, the study was a cross-sectional study. As a result, only the correlations between the levels of IL-1β, TGF-β, CRP, and MMSE-K, CDR, GDS, GDS-K scores in the three groups categorized as cognitive function were obtained. Therefore, longer-term prospective studies are needed to confirm causality. Second, the sample size was relatively small, so it is still not enough to draw a clear conclusion. A more extensive research on the community will be needed in the future. Third, there was a lack of data on nutritional status, dietary intake, overall exercise, smoking, drinking, medical illness such as heart disease, asthma, diabetes mellitus (DM), chronic obstructive pulmonary disease (COPD) that could affect inflammatory factors.
This study was a cross-sectional, only the association of the three groups with the results of hematologic tests for IL-1β, TGF-β, and CRP were known. Therefore, a larger-scale longterm follow-up study is needed to confirm causality.
It is thought that there is a limit in confirming the causal relation of the three groups and hematologic test results such as those for IL-1β, TGF-β, and CRP. Further studies are needed to elucidate the underlying biological mechanisms for other inflammatory factors not studied here, such as IL-6, IL-18, IL-12, and TNF-α.
Our study is less invasive than the CSF sampling, because it samples blood. So, blood-based studies like ours are easy to gather data and will be useful for clinical practice. Also, blood-based biomarker will be more useful than CSF-based in the future.
Acknowledgements
The authors are deeply grateful for the participation of the patients and their caregivers. We also gratefully acknowledge the assistance of the staff of the Department of Psychiatric Medicine and Dementia Clinic of Inje University Ilsan Paik Hospital, Goyang, Republic of Korea.
Notes
The authors have no potential conflicts of interest to disclose.
Author Contributions
Conceptualization: Jun Kyung Park, Kang Joon Lee, Hyun Kim. Data curation: all authors. Formal analysis: Jun Kyung Park, Kang Joon Lee, Hyun Kim. Investigation: Jun Kyung Park, Kang Joon Lee, Hyun Kim. Methodology: Jun Kyung Park. Project administration: Kang Joon Lee, Hyun Kim. Resources: Jun Kyung Park. Software: Jun Kyung Park. Supervision: Kang Joon Lee, Hyun Kim. Validation: Kang Joon Lee, Hyun Kim. Visualiztion: all authors. Writing—origianl draft: Jun Kyung Park. Writing—review & editing: Jun Kyung Park, Kang Joon Lee, Hyun Kim.