Pseudotumoral Presentation of Cerebral Amyloid-Beta Angiopathy: Case Report and Review of Literature
Article information

Abstract
Objective
Cerebral amyloid angiopathy-related inflammation (CAA-RI) is a rare and potentially treatable encephalopathy that usually affects people older than 50 years old and has an acute or subacute clinical presentation characterized by rapidly evolving cognitive decline, focal deficits and seizures. In a small subset of patients the disease can adopt a pseudotumoral form in the neuroimages that represents a very difficult diagnostic challenge.
Methods
Here in we report a patient with a tumour-like presentation of histopathologically confirmed CAA-RI.
Results
We also conducted a search and reviewed the clinical and radiological features of 41 cases of pseudotumoral CAA-RI previously reported in the literature in order to identify those characteristics that should raise diagnostic suspicions of the disease, there by avoiding unnecessary surgical treatments.
Conclusion
The therapy of CAA-RI with steroids is usually effective and clinical and radiological remission can be achieved in the first month in approximately 70% of cases.
INTRODUCTION
Sporadic cerebral amyloid angiopathy (CAA) is characterized by deposition of β-amyloid in the media and adventitia layers of the small and medium-size cortical and leptmeningeal brain arteries and less frequently in veins and capillaries [1]. CAA is frequent in the elderly. Population-based studies show that the incidence of CAA increases with age: Masuda et al. [2] studied 400 autopsy cases from Hisayama, Japan and reported an incidence of CAA of 4% to 10% in people 50–59 years old and that rises to 42% to 45.8% in those 90 years or older. A more recent autopsy study [3] performed in 404 community-dwelling persons of an average age at death of 86.5 years old found CAA was present in almost all cases with dementia (94%) and in most of those without (77%), while only 1/5 had moderate to severe disease, which is in accordance with previous reports [4]. In Alzheimer’s disease specifically the prevalence reaches 80–90% [2,5]. Classic clinical presentation includes brain hemorrhages (lobar intracerebral hematoma, cortical microhemorrhage, focal convexity subarachnoid hemorrhage and cortical superficial siderosis) and ischemic lesions (cortical microinfarctions and white matter ischemic changes) [2,6]. In a subset of patients, usually younger, there is inflammation related to CAA that can encompass a range of involvement from only a perivascular lymphocytic infiltrate to a transmural granulomatous destructive angiitis [1]. Recent reports label this spectrum of inflammatory changes as cerebral amyloid angiopathy-related inflammation (CAA-RI) [7], which is the term we choose to employ. Clinical syndrome of CAA-RI manifests as subacute cognitive decline, seizures, focal deficits and head aches [1,7-10]. These clinical symptoms correlate with asymmetric T2-hyperintense leukoencephalopathy on MRI that is responsive to immunosuppressive treatment [1,7-9]. In rare instances, CAA-RI presents as an infiltrative mass-like lesion mimicking a tumor [10]. This variant represents a diagnostic challenge and should be recognized to avoid unnecessary surgical procedures. We report a patient with CAA-RI resembling a brain tumor and review similar cases reported in the literature.
CASE PRESENTATION
A 54-year-old right-handed man developed acute aphasia and confusion followed by a generalized tonic clonic seizure. His past medical history was remarkable for high blood pressure, smoking, chronic obstructive pulmonar disease, depression and a Bell’s palsy three years previous to the event. His daily medication consisted of enalapril and clonazepam. Neurological examination revealed non-fluent aphasia. A brain MRI disclosed a left temporal lesion with hyperintense signal in T2-weighted and FLAIR sequences without contrast enhancement. Diffusion weighted images of temporal lesions did not show restricted diffusivity. Review of T2* weighted gradient echo sequence (GRE) disclosed a few cortical microhemorrhages inside the temporal lesion (Figure 1A-E). A proton MRI spectroscopy of the left lesion revealed no remarkable data. A 11C-PiB PET revealed extensive and bilateral brain cortical deposition of β-amyloid (Figure 1F). CSF exam was normal and the CSF viral polymerase chain reactions including herpes simplex virus type 1 and 2, varicella-zoster virus and enterovirus were negative. The patient was treated with valproic acid and dexamethasone. Laboratory studies including HIV serology, VDRL, eritrosedimentation rate, collagen disease tests and thyroid and paraneoplastic antibodies were negative. An electroencephalogram showed slow left temporal waves. A stereotaxic biopsy revealed pleomorphic and disposition of abigarrated glial cells suggestive of glioma. Resection of the left temporal lesion identified that cortical and leptomeningeal small and medium-sized arterial walls were thickened by an amorphous eosinophilic PAS positive substance that partially or completely occluded the lumen of the vessels. Immunolabeling of arterial wall deposits with β-amyloid antibodies was strongly positive (Figure 2). Eosinophilic thickening of the parietal wall with peri adventitial hemosiderin deposits (old microhemorrhage) was disclosed. Steroids were gradually tapered. The patient has remained symptom-free after 24 months of follow up and has returned to work.
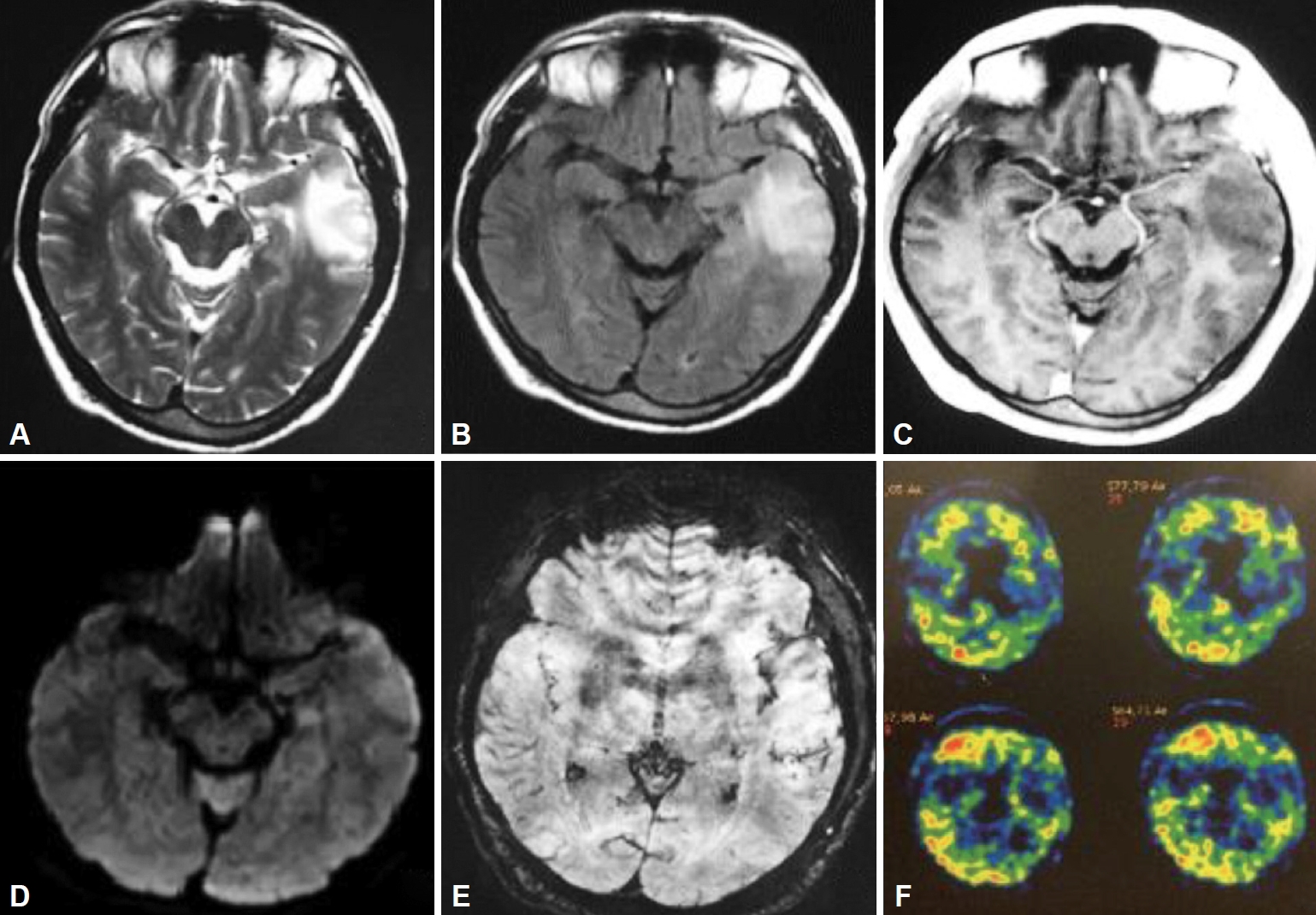
Left anterior temporal lesion with mass effect shows high signal in T2 W (A) and FLAIR (B) and hypointensity in T1W, with lack of contrast enhancement (C). High signal in DWI sequence (D) and ADC map (not shown) is due to vasogenic edema. Scattered intralesional foci of microhemorrhages can be seen in SWI (E). C-PiB PET: Extensive and bilateral cortical deposits of Beta amyloid (F). MRI: magnetic resonance imaging, DWI: diffusion-weighted magnetic resonance imaging, FLAIR: T2-weighted-Fluid-Attenuated Inversion Recovery, ADC map: apparent diffusion coefficient, C-PiB PET: C-Pittsburgh compound B Positron Emission Tomography.
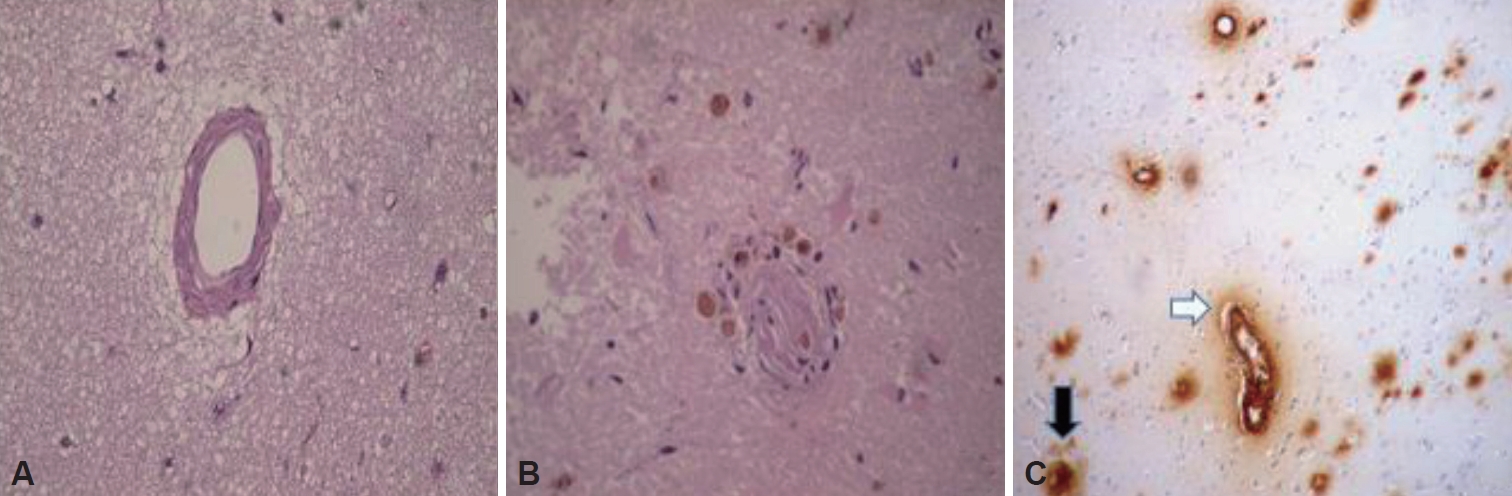
Brain biopsy, hematoxylin-eosin stain where it is observed. A: Homogeneous eosinophilic thickening of the vascular wall. B: Eosinophilic thickening of the parietal wall with peri adventitial hemosiderin deposits (old microhemorrhage). C: Positive immune staining of β amyloid in the vascular walls (black arrow) and in plaques within the parenchyma.
LITERATURE REVIEW-METHODS
We searched in Pubmed for relevant articles dating from January 1970 to January 2017 using the following key words: “cerebral amyloid angiopathy” and “pseudotumoral,” “tumor-like,” “neoplasm,” or “mass effect.” We decided to include only cases with MRI evaluation and pathologically proven diagnosis of CAA with or without related inflammation, as well as those that met diagnostic criteria proposed by Chung et al. [7] for probable CAA-RI. All relevant articles were retrieved and checked. We found 27 publications that met inclusion criteria, from which information was extracted for 41 patients. We added our patient, and data was analyzed for age, gender, clinical presentation, brain MRI findings, treatment received and evolution for the 42 cases as a group.
RESULTS
Our patient presented sudden aphasia and seizures secondary to a tumor-like lesion that resembled a low grade glioma but that finally corresponded to pathology-confirmed CAA-RI. This tumefactive mass-like aspect has been described in the literature in 15% of CAA-RI and 14% of CAA without inflammation [1]. Most of these latter are probably inflammatory forms of CAA, but a patchy distribution of perivascular lymphocytic infiltrates [7], or the possible disappearance of inflammatory infiltrates due to onset of steroid treatment before the performance of cerebral biopsy, could lead to misdiagnosis of CAA-RI as without inflammation [11]. In a more recent review of CAA-RI, Danve et al. [9] reported that up to 26% of CAA-RI cases had mass-like lesions that were usually asymmetrical and either non-enhancing or minimally enhancing. The most frequent brain malignancies suspected in cases of pseudotumoral presentation of CAA-RI were low grade gliomas [12-20], lymphomas [21,22], multifocal glioma [23,24], oligodendroglioma [25], gliomatosis cerebrii [26] and metastases [27]. The pseudotumoral form of CAA-RI can be a challenging diagnosis, as its clinical and radiological presentation is not specific [10]. The acute onset of focal neurological deficits in a patient older than 50 years can be initially misdiagnosed as an acute stroke [28], especially if the patient has vascular risk factors, as had occurred in our case. However, acute clinical presentation is not infrequent in CAARI patients, as it has been described in up to 54% of them [9], including aphasia in 16.7% [8] to 26% [7] of cases, and seizures in 31% [7] to 36% [8]. These percentages are similar for the pseudotumoral form of the disease according to our review of 42 cases from the literature: aphasia in 10/42 (23.8%) and seizures in 16/42 (38%). However, subacute cognitive decline is the most frequent clinical presentation in our series 29/42 (69%), as in others [10,19,29-31]. Therefore, CAA-RI and its pseudotumoral variant should also be taken into account as a differential diagnosis in any case of rapidly evolving dementia [32,33]. Headache was present in only 9/42 (21.4%), and 62% of patients in our review presented more than one neurological symptom (Table 1).

Clinical, imaging and histopathological findings in 42 patients with pseudotumoral presentation of amyloid angiopathy
Pathogenesis of CAA-RI is not clear. CAA-RI appears to be caused by an autoimmune response to Aβ amyloid. The autoimmune mechanism of the disease is supported by the finding of autoantibodies against beta amyloid 1–40 and 1–42 in blood [34] and CSF [35,36] of patients with CAA-RI. DiFrancesco et al. [36] reported higher titers of autoantibodies against β- amyloid 1–40 and 1–42 in CSF of a patient with CAA-RI compared with age-matched controls that, interestingly, decreased in response to steroid treatment. These autoantibodies might serve as a biomarker of the disease and allow improvement in diagnosis and monitorization of therapeutic response. It remains unclear whether inflammation is triggered by Aβ amyloid or its associated components like ApoE [9]. There seems to be a strong association of CAA-RI with ApoE genotype ε4/ε4, as approximately 76% of patients affected by the disease are homozygotic for this genotype, while only 5% of subjects with pathologically confirmed noninflammatory CAA present it [odds ratio (OR) 61.7, 95% CI 7.2 to 706, p<0.0001] [30]. We could not perform these tests in our patient.
Brain MRI in CAA-RI cases usually shows a distinctive pattern of asymmetric confluent T-2 hyperintense lesions extending through the cortical and subcortical regions with signal suggestive of vasogenic edema and the presence of multiple cortical or subcortical microbleeds in T2*GRE/SWI with variable and patchy leptomeningeal and/or parenchymal contrast enhancement [30]. Although definitive diagnosis of CAA-RI is still histological, Kinnecom et al. [30] suggested that a diagnosis of probable CAA-RI might be made on the basis of typical clinical and radiological findings without the performance of a brain biopsy. MRI diagnostic criteria for probable CAA-RI was then proposed by Chung et al. [7] and recently modified and validated by Auriel et al. [37] (Table 2).

This diagnostic criteria for probable CAA-RI yields a diagnostic sensitivity and specificity of 82% and 97%, respectively [37]. MRI in our patient showed an isolated lesion that was atypical because of its pseudotumoral aspect and the scarcity of cortical microhemorrhages. This latter was a confounding factor in our case: despite performing susceptibility-weighted imaging (SWI) sequences, brain MRI disclosed only a few gathering intralesional microhemorrhage foci that gliomas usually present [38] and not the multiple cortical or subcortical microhemorrhages inside and outside the lesion that are commonly described in cases of CAA-RI. Other authors have also found the absence of this cortical microhemorrhage in 13% [9] to 59% [8] of cases. This absence therefore does not discard the diagnosis of CAA-RI. However, as microbleeds can be missed if T2* weighted GRE or other SWI are not performed, we do recommend the inclusion of these sequences as part of MRI protocol to evaluate patients with tumor-like lesions. Only 17 of 42 patients in our series had T2*GRE /SWI sequences, and microbleeds were present in 16 (94%) of them [38].
DISCUSSION
Our patient underwent an open brain biopsy immediately previous to the surgical resection of the brain mass. Although cortical and leptomeningeal tissue was obtained, as it is required to achieve histologic confirmation [10], brain biopsy was non-diagnostic in our patient probably because adequate techniques to detect Aβ amyloid were not performed due to lack of diagnostic suspicion. After the surgery, immunolabelling of resected tissue vessels with Aβ amyloid antibodies confirmed amyloid angiopathy, and C-PiB PET imaging revealed extensive Aβ amyloidotic cortical deposits in spite of absence of microhemorrhages in those localizations. MR spectroscopy of the lesion did not reveal any increment of cholina in this case and was unremarkable in the only 7 patients of our series that underwent this technique.
Treatment of CAA-RI with steroids is usually effective, and clinical and radiological remission can be achieved in the first 3 weeks in approximately 70% of cases [7,9]. Many patients remain symptom-free after several years of follow up [10,13,14,25] but relapses can occur in 26% of cases [9], generally after reduction or cessation of immunosuppression [20,27,39] therefore, clinical surveillance is necessary, and cyclophosphamide or azathioprine can be added in relapsing disease [20,39]. In the present pseudotumoral CAA-RI series, 27/42 patients received steroids as therapy, and in 15 of these, steroids were the only pharmacological treatment; 7/27 were also treated with cyclophosphamide or methotrexate as adjuvant therapy. While a review of the literature shows that only approximately 5% of cases of CAA-RI were surgically treated [8], in this series of pseudotumoral form cases of CAA-RI 26% (11/42) of patients underwent surgical resection of the lesion due to suspicion of underlying malignancy. No treatment was delivered to 9/42 patients. The outcome was favorable (a significant or complete resolution of symptoms) in 19/22 patients treated with only pharmacological therapy, in 6/11 patients that underwent a surgical procedure, and in 5/9 patients that did not receive any specific treatment. Therefore, 71.43% of patients with pseudotumoral form of CAA-RI in this series had a good outcome. A bad outcome (ongoing deterioration or death) was evident in 14.28% of cases (1/22 patients in the steroid group, in 3/11 patients surgically treated, and in 2/9 patients in the non-treated group). Our patient has not required any further treatment after the initial course of two months of steroids, as he has been symptom-free after 24 months of follow up (Suplementary Table 1 in the online-only Data Supplement) [40-45].
CONCLUSION
CAA-RI can have an acute clinical presentation that mimics stroke and, in neuroimages, can resemble a tumour [31]. Differential diagnoses must be done with low grade gliomas and CNS lymphoma. Brain MRI T2 GRE/SWI sequences should be carefully evaluated when searching for the presence of cortical microbleeds, as they suggest CAA-RI as a possible etiology; however, their absence does not rule out inflammatory amyloid angiopathy. Although in a typical clinical and radiological scenario of CAA-RI a therapeutic trial with steroids is recommended by some authors [7,30,37,39], the gold standard for the diagnosis of CAA-RI is still brain biopsy. The pathologist should be aware of clinical suspicions of CAA-RI in order to perform the specific techniques to detect Aβ amyloid.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.30773/pi.2020.0201.
Acknowledgements
None.
Notes
The authors have no potential conflicts of interest to disclose.
Author Contributions
Conceptualization: Gustavo Sevlever. Data curation: Rafael Torino. Formal analysis: Marta Ines Bala. Investigation: Migue Saucedo. Methodology: Lucrecia Bandeo. Project administration: Luciana Leon Cejas. Resources: Sol Pacha. Supervision: Pablo Bonardo. Validation: Pablo Dezanzo. Visualization: Carlos Rugilo. Writing—original draft: Claudia Uribe Roca, Fabio Maximiliano Gonzalez. Writing—review & editing: Manuel Fernandez Pardal, Ricardo Reisin.