Genome-Wide Association Scan of Korean Autism Spectrum Disorders with Language Delay: A Preliminary Study
Article information
Abstract
Objective
Communication problems are a prevalent symptom of autism spectrum disorders (ASDs), which have a genetic background. Although several genome-wide studies on ASD have suggested a number of candidate genes, few studies have reported the association or linkage of specific endophenotypes to ASDs.
Methods
Forty-two Korean ASD patients who showed a language delay were enrolled in this study with their parents. We performed a genome-wide scan by using the Affymetrix SNP Array 5.0 platform to identify candidate genes responsible for language delay in ASDs.
Results
We detected candidate single-nucleotide polymorphisms (SNPs) in chromosome 11, rs11212733 (p-value=9.76×10-6) and rs7125479 (p-value=1.48×10-4), as a marker of language delay in ASD using the transmission disequilibrium test and multifactor dimensionality reduction test.
Conclusion
Although our results suggest that several SNPs are associated with language delay in ASD, rs11212733 we were not able to observe any significant results after correction of multiple comparisons. This may imply that more samples may be required to identify genes associated with language delay in ASD.
INTRODUCTION
Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterized by disturbances with a wide range of severity in 3 domains: socialization, communication, and the presence of restricted and repetitive patterns of behavior or interest.1 Although family and twin studies have strongly suggested that autism has high heritability, there is no consensus on the underlying genetic architecture.2,3 Both high genetic and phenotypic heterogeneity of autism may be complicating factors for the identification of candidate loci, and several genome-wide screens of multiplex families have been performed to identify possible candidate regions.4-7
Autism phenotypes that are associated with 1 of the 3 core domains of autism are potential candidates for genetic mapping because they may be controlled by few loci with large genetic effects. Autism is often perceived as a spectrum disorder composed of several dimensions. Some quantitative autism subphenotypes have been suggested to be suitable for genetic studies.8,9 The heritability of these autism phenotypes has been shown by direct linkage analyses of the traits. These analyses also provided evidence for the genetic heterogeneity of ASDs. Substantial language delay, defined as a delay in the age of the first spoken word or the first spoken phrase, has been reported as one essential component of the endophenotypes of autism. In addition, it was reported that first- and second-degree relatives in autism families have more language-related problems than corresponding relatives from patients with Down syndrome.11,12 Recently, investigators conducted several linkage and association analyses using these autism endophenotypes as covariates to increase both the genetic and phenotypic homogeneity of the ASD-affected sample. They stratified affected families according to the proband's language difficulties.12,13 In this preliminary genome-wide, family-based association study, we used a transmission disequilibrium test (TDT) to narrow the range of subjects with autism (male, >4 years of age, with significant language delay) to define a more homogenous subgroup.
METHODS
Subjects
Subjects with ASD and their biological parents were recruited through the Korean Autism Research Consortium. Each child was initially screened for ASD by 2 board-certified child psychiatrists using the Diagnostic and Statistical Manual of Mental Disorders diagnostic criteria. To confirm the diagnosis, all subjects were evaluated on the basis of the Korean version of the Autism Diagnostic Observation Schedule (K-ADOS) and the Autism Diagnostic Interview-Revised (K-ADI-R). All subjects met the diagnostic criteria of ASD.14,15 In addition, all subjects included in this study were not verbally fluent in simple conversational tasks (as expected at age >4 years), and the K-ADOS module 1 was applied. In the K-ADI-R, parents reported that their children showed word and/or phrase delay during development (>24 months and >33 months, respectively).
The psychometric properties of the probands were evaluated using the Korean Educational Developmental Institute-Wechsler Intelligence Scale for Children or the Korean version of the Vineland Social Maturity Scale (K-VSMS) depending on the capacity of the children.16,17 We performed physical and neurological examinations, including electroencephalography (EEG) and chromosomal analyses, to reveal any physical or neurological conditions. Subjects who were diagnosed with neurofibromatosis, metabolic encephalopathy, organic brain diseases, fragile X syndrome, tuberous sclerosis, or those who were diagnosed with chromosomal abnormalities or other medical conditions that might be associated with ASD were excluded from the analysis. We received written informed consent from the parents. This study was approved by the institutional review boards of the institutions where the study was performed.
Genotyping
Genomic DNA was purified from whole blood samples using the FUJIFILM DNA Whole Blood Kit S and QuickGene-810. Concentration and purity analyses were performed for all samples using a NanoDrop ND-1000 spectrophotometer, and the integrity of the samples was tested by electrophoresis on a 1% agarose gel. The 260/280 optical density ratio of the samples had to be higher than 1.8 and the 260/230 ratio had to be higher than 2.0 for the samples to be included in the genotyping analyses. DNA aliquots (500 ng) were then prepared at a concentration of 50 ng/µL in a total volume of 10 µL.
After the samples were determined to be within the defined range, they were run on the Affymetrix Genome Wide 5.0, scanned, and analyzed. Array images were acquired using a GeneChip Scanner 7G with an autoloader that scanned each array. Raw DAT image files were generated using the GeneChip Operating System (GCOS) software. Each DAT image was processed by the GCOS software to generate a feature-extracted .CEL file.
All .CEL files were subjected to low-level quality control (QC) analysis using the Genotyping Console 2.1 software (Affymetrix) to determine their suitability for genotyping. This QC analysis included assessment of image quality to ensure that it was free of manufacturing or physical defects. Next, we examined the QC call rate (generated automatically when the .CEL files were imported into the Genotyping Console) of approximately 3,022 single-nucleotide polymorphisms (SNPs). The analysis of these SNPs has been reported to be sensitive to the DNA quality. This step included separate assessments of the QC call rates for SNPs that were examined in the NspI and StyI fragments. We only performed genotyping analysis on .CEL files with overall and fragment-specific QC call rates that exceeded 86%. Arrays that passed these criteria were subjected to Bayesian Robust Linear Modeling using Mahalanobis Distance genome-wide genotyping by using the Genotyping Console 2.1 at a confidence threshold score of 0.05. The mean value for the sample call rate was 98.1%. The IBS score was 1.57±0.55. Graphical representation of relationship errors (GRR), a graphical tool for verifying assumed relationships between the individuals in genetic studies, was used to detect common errors when using genotypes from many markers.18 SNPs were also subject to QC before analysis. To minimize genotyping errors, we excluded SNPs with a p value of <10-4 from the calculation of the Hardy-Weinberg equilibrium and minor allele frequencies below 1% from the analysis when using PLINK 1.0.4 (http://pngu.mgh.harvard.edu/~purcell/plink/). After drawing Q-Q plots based on a call rate between 90% and 99%, we selected a call rate of 95% to control the marker quality. After application of the QC filters, 331095 out of 440094 SNPs remained.
Statistical analyses
We determined the Mendelian inheritance error and tested the family-based association for each individual polymorphism using the standard TDT method. In addition, we used MDR-PDT to detect epistasis on a genome-wide scale with 194 markers that had a p-value of <10-3 in the TDT test.19 The false-discovery rate (FDR) is a method that considers the expected proportion of significant tests that are truly null. The FDR procedures proposed by Benjamini and Hochberg20 were applied to adjust for multiple comparisons.
RESULTS
Clinical features of patients with autistic spectrum disorders
The average age of the 42 probands was 77.7±22.6 months (mean±SD; range, 49-149 months). All probands were males. The social quotient measured using the K-VSMS was 50.5±14.8 (range, 23-72 months). The mean IQ score, available only for 9 subjects because of the low level of functioning, was 46.2±12.2 (range, 31-65). The K-CARS score was 33.3±4.4 (range, 23-46). The average age at which the children spoke their first words, as reported by the parents in the K-ADI-R, was 35.9±21.6 months (range, 10-85 months). The age at which the children spoke their first phrase was 58.1±21.4 months (range, 13-108 months). Thirteen patients (30.6%) had EEG abnormalities suggesting a partial seizure or diffuse cerebral dysfunction, but none were diagnosed with a clinically significant partial seizure disorder.
Nine subjects (21.4%) had not yet spoken any significant meaningful single words, whereas another 13 subjects (30.6%) were not able to say developed 2-word phrases, including verbs, in spite of their ability to use 5 different single words every day. The mean communication domain score on K-ADOS was 6.0±1.5, and the score for the qualitative abnormalities in the communication domain on K-ADI-R was 14.4±3.1. All subjects obtained scores that exceeded the cut-offs. The subdomain score for the lack of/delay in spoken language and failure to compensate through gestures in the nonverbal subjects was 7.4±1.1, and the score for the lack of varied spontaneous make-believe or social imitative play was 5.7±0.7. All subjects showed significant disturbances in social interaction, repetitive behavior, and restricted interest domain based on the diagnostic algorithms of both K-ADOS and K-ADI-R.
Association analysis results
The distribution of p values for the TDT is shown in Table 1. The distribution of p values examined in the discovery dataset was found to be closely matched to that expected for a null distribution, except at the extreme tail of low p values (Figure 1). The results of the family-based genome-wide association analyses are presented in Figure 2. The 30 most powerful properties according to the TDT results are shown in Table 2. The most statistically significant association before correction of multiple comparisons was found for an SNP (rs11212733) on chromosome 11q22.3 (p-value=9.76×10-6).
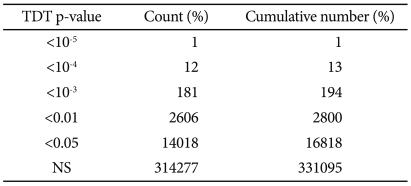
The distribution of p values for TDT
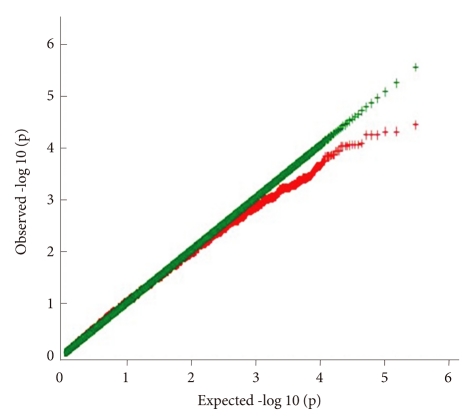
Quantile-Quantile (Q-Q) plot of TDT p-value. TDT: transmission disequilibrium test.
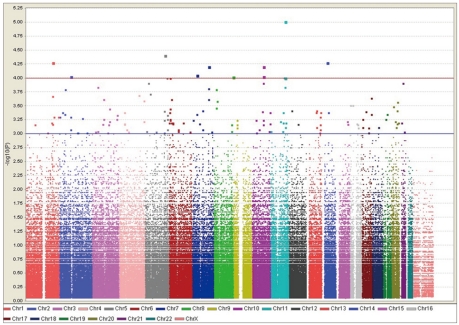
Genome wide family-based association of Korean ASDs with language delay. Results of TDT (-log p-value) for 331095 SNPs plotted in chromosomal order. ASDs: Autism spectrum disorders, TDT: transmission disequilibrium test, SNPs: single-nucleotide polymorphisms.
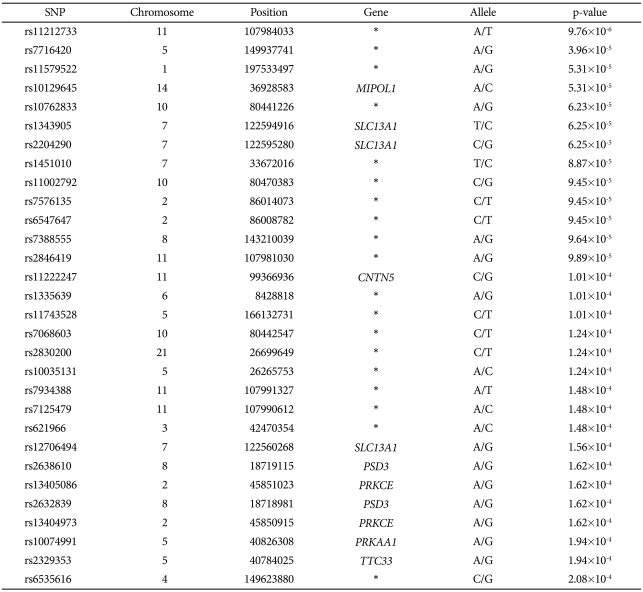
The top 30 hits from the TDT results
In the MDR-PDT analysis, we detected best models from 1-locus to 4-locus. The results of the MDR-PDT are presented in Table 3 and Figure 3. In particular, rs7125479, which is also located in chromosome 11, was contained 1-locus, 2-locus, and 4-locus model. We also found that rs7125479 and rs11212733, the most significant SNPs in this study, were in linkage disequilibrium (r2=1.0) in the HapMap database. After correction for multiple comparisons using FDR, none of the SNPs remained significant.
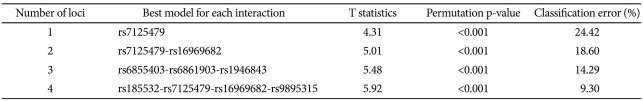
Summay of MDR-PDT results
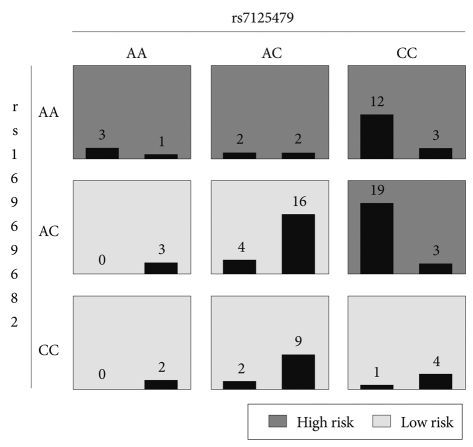
MDR-PDT 2-locus model of rs7125479 and rs16969682. Each multifactorial cell is labeled as "high risk" or "low risk". For each multilocus combination, empirical distributions of affected (left bar in cell) and unaffected (right bar in cell) are shown.
DISCUSSION
When we applied the 400 kb sequence of chromosome 11q-22.3 to the International HapMap database, we observed that rs11212733 is located in the 5' region of the exophilin 5 gene and DEAD (Asp-Glu-Ala-Asp) box polypeptide 10 gene (DDX10). Yonan et al.7 provided evidence suggesting a linkage of chromosome 11 and others to ASDs in a genome-wide screen, which was conducted to identify autism susceptibility in the loci of 345 families. In addition, Schellenberg et al.5 reported a strong linkage for chromosome 11, which was unique to the male members of ASD families. With regard to the phenotypes of ASDs, there is a report on the quantitative social responsiveness scale genome scan, which identified a locus on chromosome 11.21 Liu et al.22 suggested a possible linkage of chromosome 11q15 to ASD based on a genome-wide linkage analysis of quantitative and categorical autism subphenotypes in ASD families with a delayed onset of speaking their first phrases. Although statistically not significant after correction for FDR, our results corresponded with previous results, which revealed a possible association of chromosome 11 with autism.
In a quantitative linkage scan for a language endophenotype in autism, Alarcón et al.23,24 identified a quantitative trait locus (QTL) related to language delay across a 10 cM region on chromosome 7q35, and this evidence was supported by a follow-up study. Recently, the contactin-associated protein-like 2 gene, a member of the neurexin family, which is located on chromosome 7q35, has been shown to be a language-related autism QTL. The authors suggested a strong a priori candidate gene of autism with significant association results.25,26 However, we failed to replicate those results. Only 3 SNPs (rs1343905, rs2204290, and rs12706494) located on chromosome 7q32 (SLC13A1 gene) were among the 30 SNPs that had the most powerful properties in the TDT.
The general goal for genome-wide association studies of complex disorders is to find multiple genes with a small effect; however, the statistical power of our sample was not high enough to confirm an association with SNPs that may have very small genetic effects at the population level (odds ratios <1.4).27 This negative output might be due to the small sample size and is not surprising given the recent results of other studies for complex psychiatric disorders such as attention deficit hyperactivity disorder and bipolar disorder.27,28 Recently, to solve the problem of low statistical power, several genome-wide association studies were conducted in a large number of ASD families, with a combined sample set of more than 10,000 subjects of European ancestry.6
Despite the small sample size and lack of significance after correction for multiple comparisons, this study is the first genome-wide scan in an Asian population with ASD. Our samples, in particular, represent a homogenous subset, i.e., males over 48 months of age with a significant language delay. As ASD is one of the heterogeneous phenotypes among psychiatric illnesses, extracting a homogenous subgroup may enhance the power of detecting genetic effects.29 Usable traits vary from one autistic person to another. The frequency of milder forms in nonautistic family members is significantly higher than in controls, and in particular aggregation in autism families.30 Language delay is 1 plausible trait that is frequently presented in siblings and first-degree relatives of children with ASD.31 In this study, we detected candidate SNPs in chromosome 11, rs7125479, and rs11212733 as markers of language delay in ASD. Future replication with a larger sample size and sufficient statistical power may be required to identify a significant association and confirm significant results.
Acknowledgments
This study was supported by a grant of the Korea Healthcare technology R&D project, Ministry of Health & Welfare, Republic of Korea (A080651) and Mid-career Research Program through NRF grant funded by the MEST (2010-0007583).