INTRODUCTION
Electroconvulsive therapy (ECT) is a treatment modality with a long historical background, and has been increasingly applied to various neuropsychiatric problems [1,2]. Major applications of ECT are major depressive disorder, catatonia, schizophrenia, and bipolar disorders, but recent advances have made them to be extended to various other neuropsychiatric conditions, such as Alzheimer dementia, behavioral problems of autism spectrum disorders, self-mutilating behaviors as well as Parkinson disease (PD) and intractable seizure [3-6].
The beneficial effects of ECT on PD have been reported repeatedly by, mainly, case studies. ECT can assist the improvement of neuropsychiatric symptoms, including depressive mood and psychotic disorders, related to PD [7-9]. Moreover, it has been demonstrated that ECT is able to ameliorate the motor deficits itselfâthe core symptom of PD [9-11]. With these foundations, ECT has been suggested as a promising treatment option, especially in case of patients having both motor deficits and psychiatric problems that are difficult to be controlled by medication [10]. However, the effectiveness of ECT on PD requires further confirmation, and the underlying mechanisms have not been investigated yet.
Animal models of neuropsychiatric disease could be an efficient tool to provide evidence of therapeutic effectiveness of ECT as well as to search for the underlying mechanism of its action in brain which is hardly accessible in human patients. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride (MPTP) is a neurotoxin which selectively destroys dopaminergic (DA) neurons in the substantia nigra compacta (SNc) [12-14]. Systemic injection of MPTP in mice induces behavioral, cognitive and neurochemical changes resembling PD, which is one of the mostly-utilized PD animal model [14-17].
A further point to note is that although motor deficits are considered as a primary diagnostic and therapeutic target of PD, recent studies have emphasized the significance of non-motor symptoms, such as cognitive, emotional, and perceptual deficits, in the disease [18-20]. The progression of non-motor symptoms involves several brain regions and neurotransmitters. The deficiency of dopamine and its transporter in striatum, which receives dopaminergic projections from SNc, has been linked to depression, cognitive deficit, and dementia in PD [18]. Notable atrophy of prefrontal cortex (PFC) and consequent impairment of functional connectivity in the region were evident in PD patients with dementia [21]. In addition, dopaminergic, noradrenergic, and serotonergic alterations were observed both in the striatum and the PFC in early phase of PD accompanying emotional and cognitive deficits [22,23]. Interestingly, a MPTP-treated macaque model study observed that specific transcriptomic changes took place outside the basal ganglia structures even before motor symptoms became apparent, and an appreciable part of the transcriptomic changes were also observed in the PFC of the post-mortem samples of patients, both of which again strengthen the notion that non-motor symptoms and its underlying causes may be deeply involved in the progression of PD [24].
In the present study, we examined the effects of electroconvulsive seizure (ECS), an animal model for ECT, on the MPTP-induced Parkinson model mice and its underlying mechanisms in the brain. ECS has been reported to induce cellular proliferation, synaptic modifications, and the upregulation of neurotrophic factors [25]. Moreover, our recent studies reported that repeated ECS treatments induced autophagy activation and more autophagosome formations in the rat frontal cortex via AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signaling pathway [26,27]. Autophagy impairments in the neurodegenerative disorders such as PD and Alzheimer disease have been increasingly given attention, and autophagy manipulation is currently highlighted as a novel treatment strategy for the diseases [28,29].
Here, we demonstrated that repeated ECS treatments normalized motor deficits in the MPTP PD mouse model. The treatment also prevented the MPTP-induced DA neuronal loss in SNc, and ameliorated autophagy dysfunctions in the PFC of the mouse model.
METHODS
Animals
The animals were treated in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023), and formal approval to conduct this experiment was obtained from the Animal Subjects Review Board of Dongguk University Hospital and Seoul National University Hospital. Male C57BL/6J mice (150-200 g, 8 weeks old) were housed for 1 week before the experiments and maintained under a 12-h light/12-h dark cycle (light on 8:00 AM) with food and water available ad libitum.
MPTP and ECS treatments
MPTP (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in saline. The mice received four intraperitoneal (i.p.) injections of saline or MPTP at a dose of 20 mg/kg at 2 h intervals according to the previously validated method [14,30].
From 7 days after MPTP or saline injection, ECS or sham treatment was administered to the mice 3 times per week for 2 weeks at the same time (12:00-13:00) for a total of 6 times. After the last ECS or sham treatment, rotarod test was performed, and the mice were decapitated 24 h after the last treatment, and PFC, striatum, and midbrain were dissected. The mice were decapitated 24 h after the last treatment, and their brains were dissected.
ECS was administered to the mice via ear-clip electrodes and a pulse generator (UgoBasile ECT Unit-57800-001; UgoBasile, VA, Italy) at a frequency 60 pulses/s, with a pulse width of 0.4 ms, shock duration of 0.4 s, and current of 55 mA. Shamtreated control animals were handled in the same fashion as the ECS treatment group, but no electric current was delivered. An ECS-induced seizure was validated by observing general convulsions consisting of tonic and clonic phases and measuring the duration of the convulsions. ECS-treated mice that underwent a generalized convulsion for more than 30 s were included for further analysis. To prevent death due to respiratory failure, mice were placed with the plastic cage filled with oxygen before and after ECS until they began breathing regularly as done in the previous report [31].
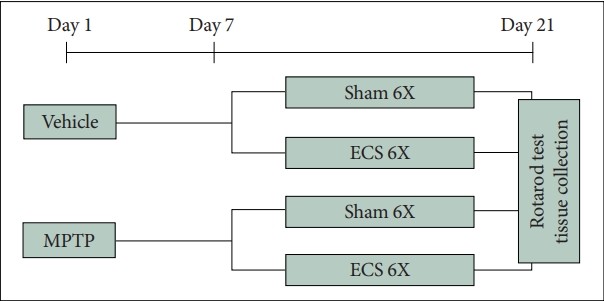
Mice received MPTP on day 1 and ECS treatments from day 7. The ECS was delivered total 6 times for 2 weeks. On day 21, 24 h after the last ECS treatment, rotarod test was performed and brains of the mice were collected for molecular analysis. Mice were divided into four groups: vehicle & shamtreated group (VS), vehicle & ECS-treated group (VE), MPTP & sham-treated group (MS), and MPTP & ECS-treated group (ME) (Figure 1).
Rotarod test
Rotarod test was performed based on the previously validated methods [32] with minor modifications. All animals were subjected to the rotarod test by a blinded investigator. On the testing day, animals were first pre-trained on the rotarod apparatus (UgoBasile) 1 h before the main trial. Mice were located on a rotating rod accelerating from 5 to 20 rpm for a 300 s time period. The results were recorded as the time in seconds at which the mice fell off from the rung of the rod, and the mice that completed the task over 300 s scored as 300.
Immunoblot analysis
The dissected brain tissues were homogenized as described previously [33,34]. After centrifugation at 20,000 à g for 20 min at 4°C, the supernatants were boiled in Laemmli sample buffer. The immunoblot analysis was performed as described previously [33,34]. The membranes were incubated with primary antibodies overnight at 4°C, followed by a second incubation with anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (HRP; Jackson Immuno Research Laboratories Inc., West Grove, PA, USA). We used the following primary antibodies specific for the following molecules, which are presented with their molecular weight, Research Resource Identifier, and dilution ratio, respectively. Primary antibodies specific for β-actin (42 kDa, AB_330331, 1:10,000), LC3A/B (14, 16 kDa, AB_2137703, 1:1,000), AMPKι (62 kDa, AB_330331, 1:1,000), p-Thr172-AMPKι (280 kDa, AB_2219397, 1:1,000), Unc-51-like kinase 1 (ULK1; 161 kDa, AB_2214706, 1:1,000), p-Ser317-ULK1 (140-150 kDa, AB_2687883, 1:500), Beclin1 (60 kDa, AB_879596, 1:1,000, Abcam), p-S93-Beclin1 (60 kDa, AB_2688032,1:1,000), Raptor (150 kDa, AB_561245, 1:1,000), p-Ser792-Raptor (150 kDa, AB_2249475, 1:1,000), S6 (32 kDa, AB_331355, 1:1,000), p-Ser235/236-S6 (32 kDa, AB_331679, 1:1,000) were used. The membranes were developed using the enhanced chemiluminescence system (Pierce Biotechnology, Rockford, IL, USA) and were then exposed to X-ray film (AGFA CurixRPI, Mortsel, Belgium). Immunoblot signals on developed X-ray film were quantified with the TINA program, version 2.10G (Raytest, Straubenhardt, Germany).
Immunohistochemistry
Immunohistochemistry was performed using a free-floating method. Mice were anesthetized with urethane (1.5 g/kg, i.p.) 24 h after the last ECS treatment and perfused intracardially with 0.1 M phosphate-buffered saline (PBS; pH 7.4) followed by 4% paraformaldehyde (Sigma-Aldrich) in 0.1 M PBS (pH 7.4). Brains were sectioned at 25 Îźm on a cryostat (Leitz, Wetzlar, Germany), then immediately immersed in a cryoprotectant, 50% glycerol in 0.1 M PBS. Immunohistochemistry was performed with the ABC system (Invitrogen, Waltham, MA, USA). Sections were washed with and incubated in 0.3% H2O2 for 30 min to quench endogenous peroxidase activity. After extensive washing with 0.1 M PBS, sections were blocked with 5% normal goat serum at room temperature for 30 min, then incubated overnight with primary antibody against tyrosine hydroxylase (TH) or Îą- synuclein (Cell Signaling Technology, Danvers, MA, USA) at a dilution of 1:500 at 4°C. Sections were incubated with biotinylated secondary antibodies and then incubated with HRP-conjugated streptavidin. Signals were detected using DAB substrate (Sigma-Aldrich). Subsequently, sections were mounted with DPX mountant (Fluka, Switzerland). Images of the substantia nigra compacta according to the Allen mouse brain atlas (http://mouse.brain-map.org) were obtained using an Olympus microscope connected via Leica DFC280 digital camera with an software (Leica ApÂplication Suite V3; Leica, Wetzlar, Hesse, Germany) under a 40Ă microscope objective, and all the TH-positive cells in the 600Ă800 Îźm areas of each brain regions were counted for quantitative analysis. The immunoreactivity of Îą-synuclein in substantia nigra (SN) was quantified using ImageJ (NIH, Bethesda, MD, USA).
Statistical analysis
Rotarod test results are expressed as time (sec), and the immunoblot and Îą-synuclein immunohistochemistry results are expressed as relative optical densities (ODs) (i.e., the OD percentages relative to VS control mean values). Results are presented as meanÂąstandard error of the mean (SEM). The mean rotarod test time, relative immunoreactivity ODs on the immunoblot and Îą-synuclein immunohistochemistry, and the number of immunoreactive-positive cells were compared among VS, VE, MS, and ME groups were analyzed using two-way analysis of variance (ANOVA) with respect to MPTP and the ECS treatments, and the comparison between groups was performed with post hoc Tukeyâs test. All tests were performed using SPSS 27.0 for Windows (IBM Corp., Armonk, NY, USA). A p<0.05 was considered significant.
RESULTS
Repeated ECS treatments improve motor impairment in MPTP PD mice
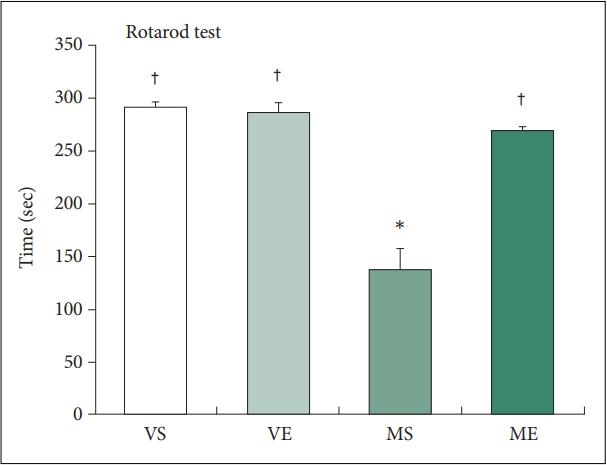
We determined whether repeated ECS treatments improved MPTP-induced motor deficits by testing motor performance by a rotarod test (Figure 2). On the test, MPTP injection significantly decreased the latency to falling to 137.8 s, a 52.9% decrease compared with the VS control group. Repeated ECS treatments nearly completely normalized the MPTP-induced motor impairment, which increased sustained rotarod time to 268.6 s. Two-way ANOVA analysis indicates that ECS (F=22.897, p<0.001) and MPTP (F=43.943, p<0.001) significantly affects rotarod time, and the effect of interaction between ECS and MPTP on rotarod time is also significant (F=27.232, p<0.001). Group comparisons using post hoc Tukeyâs tests demonstrated that rotarod time in MS group was significantly lower compared to VS group (p<0.001) and that in ME group was significantly higher compared to MS group (p<0.001). The findings clearly demonstrate that repeated ECS treatments recovered the motor deficits in the MPTP-induce PD mice model.
Repeated ECS treatments recovered DA neuron degeneration in SNc of MPTP PD mice
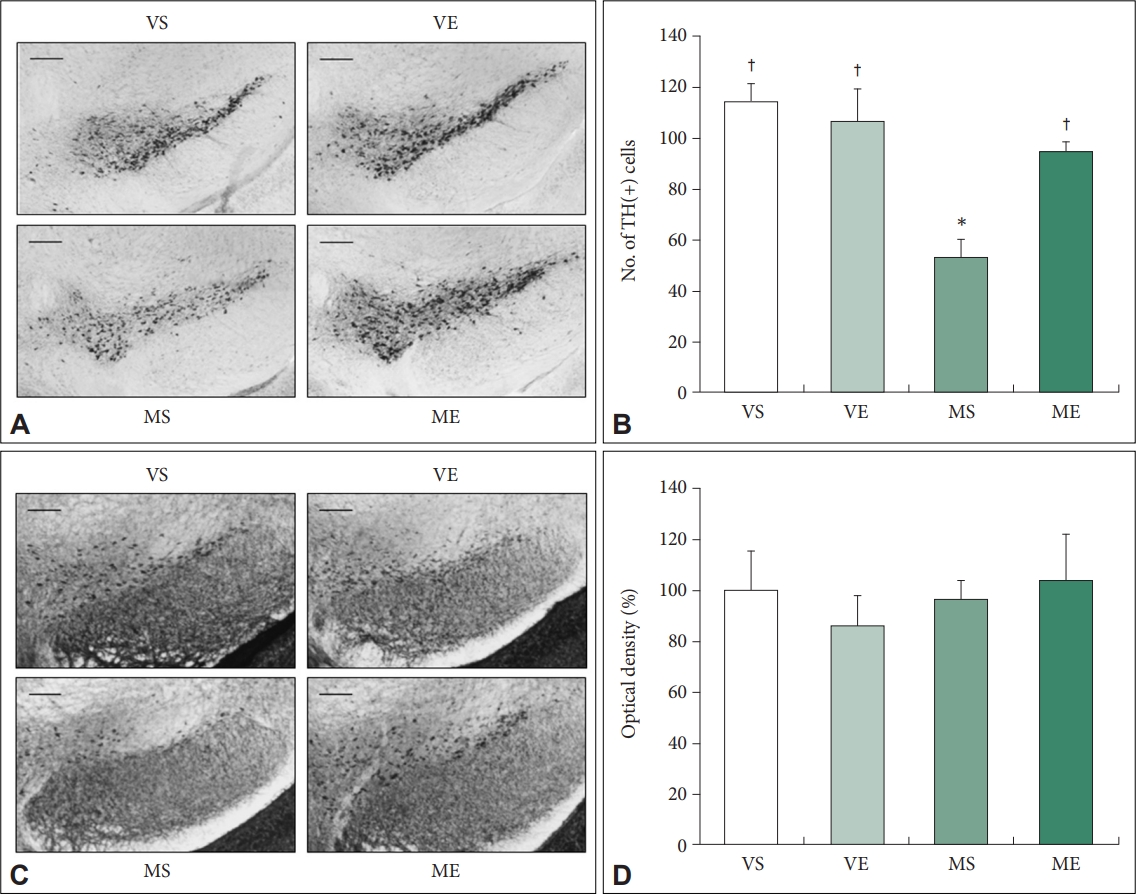
Next, we examined the effect of ECS treatment on dopamine neuron impairment induced by MPTP injection (Figure 3A and B). Number of TH-positive cells were significantly affected by MPTP (F=15.973, p=0.001) and the interaction between ECS and MPTP (F=7.118, p=0.014). MPTP treatment significant decrease of TH-positive cells in SNc compared to VS group induced a 53.6% loss of TH-positive cells in SNc. Repeated ECS treatments significantly increased the number of TH-positive neurons by 79.5% in SNc of MPTP PD mice. In MS group, TH-positive cells were significantly lower compared to VS group (p=0.006). In ME group, the number of TH-positive cells were significantly higher compared to MS group (p=0.025). The findings indicate that repeated ECS treatments prevent DA neuron degeneration in the SNc of MPTP PD mice model.
Abnormal accumulation of Îą-synuclein is one of the major pathological characteristics of PD, which may induce motor impairment if it occurs in the midbrain [35]. We examined the effect of MPTP injection and ECS treatment on the accumulation of Îą-synuclein in the SN, but could not detect any significant difference between groups (Figure 3C and D), as previous studies in MPTP PD mice model using same drug treatment paradigm with the present study have reported similar results [36,37].
Effects of repeated ECS treatments on the LC3 autophagy signaling in mice PFC, striatum, and midbrain of MPTP PD mice
We have reported that 10-daily repeated ECS treatments induce activation of autophagy via AMPK activation in the frontal cortex of rats. 26 In this study, we have examined whether the findings are also relevant in mice ECS model with different experimental condition (ECS treatments 6 times/2 weeks) in more extended brain regions, including PFC, striatum, and midbrain. The midbrain region including substantia nigra was included for immunoblot analysis as previous other studies did [38,39].
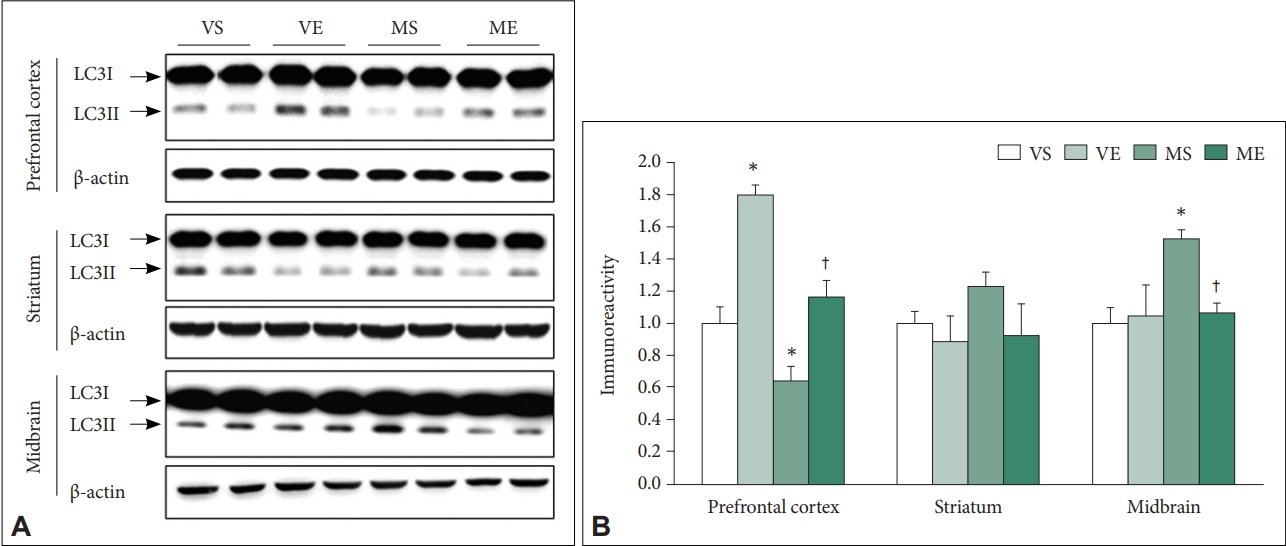
Firstly, we have determined the effects of MPTP and ECS on protein levels of LC3-II, biochemical marker of autophagy [40]. The immunoreactivity of LC3-II showed differential pattern of changes according to the examined brain regions (Figure 4). In PFC, ECS (F=32.684, p<0.001) and MPTP (F=22.212, p<0.001) significantly affected immunoreactivity of LC3-II. Group comparison demonstrated that LC3-II immunoreactivity is significantly higher in VE (p<0.001), and significantly lower in MS (p=0.038) compared to VS control group, and that in ME group was significantly higher compared to that in MS group (p=0.003). The findings indicate that, in PFC, MPTP induced impairment of autophagy signaling which is attenuated by repeated ECS treatments. There was no significant effects of ECS and MPTP on LC3-II in striatum. In the midbrain, ECS (F=12.051, p=0.002), MPTP (F=21.824, p<0.001), and the interaction between ECS and MPTP (F=9.128, p=0.004) were shown to significantly affect the LC3-II immunoreactivity. Group comparison demonstrated that LC3-II immunoreactivity was significantly higher in MS compared to VS group (p<0.001), and that in ME group was significantly lower compared to MS group (p=0.002). In midbrain, MPTP increased autophagy signaling which was reduced by repeated ECS treatments.
Effects of repeated ECS treatments on the AMPK-ULK1-Beclin1 signaling in mice PFC, striatum, andmidbrain of MPTP PD mice
AMPK is a serine-threonine kinase that plays a major role in maintaining metabolic homeostasis [41]. Activation of AMPK promotes autophagy through ULK1 [42,43], and AMPK and/or ULK1 phosphorylates and activates Beclin1, the mammalian homologue of Atg6 which initiates autophagy induction [44,45]. Previously, we have reported that AMPK/ULK1 signaling mediated ECS-induced autophagy signaling in the rat frontal cortex [26].
We have examined the activation-related phosphorylation of AMPKÎą, ULK1, and Beclin1 after MPTP and/or ECS treatments (Figure 5). In PFC, two-way ANOVA demonstrated that ECS (F=43.126, p<0.001) and MPTP (F=8.126, p=0.010) significantly affected phosphorylation of AMPKÎą. Immunoreactivity of p-Thr172-AMPKÎą was significantly higher in ME than VS (p<0.001), and was significantly higher in ME group compared to VS (p=0.042) and MS group (p=0.006). In PFC, phosphorylation of ULK1 was also significantly affected by ECS (F=30.214, p<0.001) and MPTP (F=10.022, p=0.006) and their interaction was also significant (F=6.321, p=0.032). Immunoreactivity of p-Ser317-ULK1 was significantly higher in VE (p<0.001) and ME (p=0.008) than VS group, and was significantly higher in ME than MS group (p<0.001). Beclin1 phosphorylation was significantly affected by ECS (F=28.121, p<0.001) and MPTP (F=7.121, p=0.018) in PFC. Immunoreactivity of p-Ser93-Beclin1 was significantly higher in VE (p<0.001) and ME (p=0.012) than VS group, and was significantly higher in ME than MS group (p=0.008). In striatum, phosphorylation of AMPKÎą and Beclin1 did not show significant changes in response to MPTP and/or ECS treatments, while immunoreactivity of p-Ser317-ULK1 was significantly higher in VE compared to VS group (p=0.038). In midbrain, immunoreactivity of p-Ser93-Beclin1 was significantly affected by MPTP (F=16.242, p=0.022). Immunoreactivity of p-Ser93-Beclin1 showed significant increase compared to VS group (p=0.028), and that in ME group showed significant reduction compared to MS group (p=0.044). Taken together, repeated ECS treatments was demonstrated to activate AMPK-ULK1-Beclin1 signal pathway, which was also activated in the PFC of MPTP-treated mice.
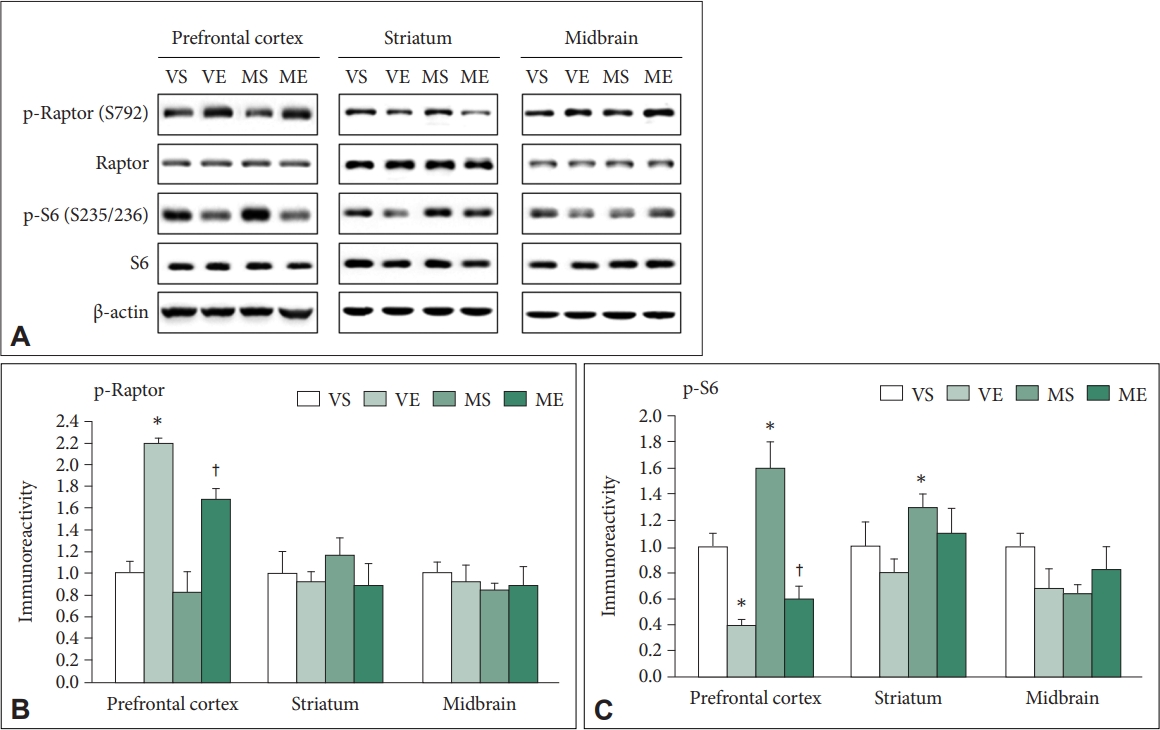
Effects of repeated ECS treatments on the Raptor and S6 in mice PFC, striatum, and midbrain of MPTP PD mice
Activation of AMPK signal pathway accompanied with inhibition of mTOR could enhance autophagy activation. Active AMPK inhibits mTORC1 by phosphorylating Raptor [46,47]. Raptor phosphorylation at Ser792 was analyzed (Figure 6A and B). ECS significantly affected the immunoreactivity of p-S792-Raptor (F=41.859, p<0.001). Immunoreactivity of p-S792-Raptor was significantly higher in VE (p<0.001) and ME (p=0.011) than VS group, and was significantly higher in ME than MS group (p=0.005). In the striatum and midbrain, p-S792-Raptor immunoreactivity did not show significant changes.
S6 protein is a major downstream effector of mTORC1 and its phosphorylation level could reflect the mTORC1 activity [48-50]. In the PFC, immunoreactivity of p-S235/236-S6 was significantly affected by ECS (F=46.224, p<0.001) and MPTP (F=5.437, p=0.031) (Figure 6A and C). Group comparison demonstrated that immunoreactivity of p-S235/236-S6 was significantly lower in VE (p=0.021), was significantly higher in MS (p=0.037) compared to VS, and it was significantly lower in ME compared to MS group (p<0.001). In striatum, p-S235/236-S6 immunoreactivity was significantly affected by ECS (F=5.665, p=0.028) and MPTP (F=7.950, p=0.011), and that in in MS group was significantly higher compared to VS group (p=0.031). In midbrain, there was no significant changes in p-S235/236-S6 immunoreactivity.
In summary, the findings suggest that repeated ECS treatments inhibits mTOR signaling in control and MPTP-treated condition, which was accompanied with activation of AMPK signaling in the mice PFC.
DISCUSSION
In this study, repeated ECS treatments markedly normalized the motor deficits in the rotarod test and prevented the loss of DA neurons, as evidenced by TH immunostaining in SNc, in the MPTP PD mouse model. In addition, the increased autophagy signaling in midbrain of the model mice, manifested by the enhancement of the LC3-II level and the phosphorylation level of Beclin-1, significantly vanished with the ECS treatments. The model mice showed, conversely, decreased LC3-II autophagy signaling in the PFC, and that was also reversed by the ECS treatments. Our molecular study demonstrated that the ECS treatments activated AMPK-ULK1-Beclin1 signaling pathway with the accompanying Raptor activation and mTOR signaling inhibition, which could promote the autophagy induction, in the mice PFC. These findings reveal the therapeutic behavioral effects of repeated ECS treatments on PD, which could be related to the neuroprotective action of ECS mediated by AMPK-autophagy signaling.
Numerous clinical reports have shown that the ECT could exert antidepressive and antipsychotic as well as motor-related anti-parkinsonian effects in PD [9,51]. To our knowledge, however, this is the first animal study demonstrating the beneficial effect of the repeated ECS treatments on the motor deficits in PD. In this study, ECS treatments repeated 6 times in 2 weeks significantly normalized the MPTP-induced motor deficits resembling those observed in PD. In line with the behavioral findings, the ECS treatments also notably inhibited the MPTP-induced loss of TH-positive cells in SNc, the region important in modulating motor function. The present study provides a heuristic animal model having predictive validity which could be utilized as the tool to examine the underlying mechanisms related to the effects of ECT on PD.
We chose PFC, striatum, and midbrain regions for immunoblot analysis. The nigrostriatal pathway connecting SNc in the midbrain with the dorsal striatum is the main pathogenic region of PD with regard to motor symptoms [52]. The frontal cortex has been associated with psychotic and cognitive symptoms of PD [53,54]. We assessed autophagy in those regions, as it is relevant to both the ECS and PD as explained in the introduction section. Autophagy process can be viewed as a double-edged sword according to its context-dependent cellular consequences: it is one of the traditional apoptotic processes inducing neuronal cell death, while its induction could promote neuroprotective action through increased clearance of neurotoxic materials [55]. Inhibition of autophagy system in SNc is one of the therapeutic strategies preventing DA neuron loss, while established autophagy activators, such as metformin or rapamycin, could enhance neuroprotection in forebrain regions in various PD animal models [56-59]. Thus, inhibition of autophagy in SNc or striatum could relate to the motor symptom treatment, while promotion of it in frontal cortex could associate with the therapeutic effects on mood, perception, and cognition. LC3 is a widely used marker for autophagy, and LC3-II, which is converted from LC3-I to initiate autophagy, is distinctly correlated with the number of autophagosomes [60]. In this study, LC3-II immunoreactivity showed the expected regional differences as it was increased in midbrain while reduced in PFC of the MPTP PD mice. Furthermore, we found that repeated ECS treatments reversed the regionspecific changes of LC3-II, which possibly represent its therapeutic capacity in PD.
We have reported that repeated ECS treatments for 10 days in rats induces autophagy via the activation of AMPK-ULK1-Beclin1 signaling 26 and AMPK-mediated mTOR inhibition [27] in the frontal cortex. AMPK plays a critical role in the autophagy process [61]. Repeated ECS treatments activated AMPK and its substrate ULK1, which increased the number of autophagosomes and the level of LC3-II and the autophagy-related (ATG) 5-ATG12 conjugate in the rat frontal cortex [26]. The activation of ULK1 and autophagy requires mTOR inhibition by AMPK activation [26]. Our previous study found that the ECS treatments increased the phosphorylation of Raptor, which is another substrate of AMPK and known to suppress mTOR activity [46], and decreased the phosphorylation of p70S6K and S6, the downstream molecules of mTOR1 [27].
To investigate those autophagy-relevant signaling changes by ECS in MPTP PD mouse model, we established our ECS protocol for mouse, 3 times per week for 2 weeks, in a trial-and-error manner, but that is very similar to the ECT protocol used in the actual human treatment. In the present study, we observed comparable results with the rat model. The ECS treatments increased the level of LC3-II and the phosphorylation of AMPK, ULK1, Beclin1, and Raptor, and inhibited S6 by dephosphorylation in PFC, suggesting ECS-induced autophagy activation. Interestingly, the phosphorylation of S6 was significantly increased in the PFC of the MPTP treated mice, which representing activated mTOR signaling, and that was inhibited by repeated ECS. In contrast to PFC, we found that the activity of most autophagy-relevant proteins in striatum and midbrain has not been appreciably changed. However, the phosphorylation level of Beclin1 was significantly high in the midbrain of the MPTP treated mice and normalized by the ECS treatments, which is consistent with the results of LC3-II immunoblotting in the region. The findings suggest that ECS-induced normalization of the reduced LC3-II level in the PFC of the MPTP PD model might be associated with AMPK-ULK1 signaling. However, ECS-induced reduction of LC3-II level in midbrain was not accompanied with the changes in AMPK and mTOR signal pathways, which indicates the involvement of other mechanisms. Further studies are required to understand the brain regiondependent different action mechanisms of ECT.
Region-specific molecular changes in the brain by ECS has been reported in previous studies. One report showed that single or 10 times ECS produced substantial transcriptional changes of unique genes in the rat brain, and the number of gene changes was three times more in the hippocampus than in the frontal cortex [62]. Another study reported that 2-day ECS in the rat evoked transcriptional changes mainly in the catecholaminergic system and the locus coeruleus of the brain stem, and of the proteins involved in synaptic plasticity in the hippocampus [63]. The underlying causes of the regional pattern and its implications in therapeutic and clinical aspects needs further investigation in future studies. In addition, although MPTP exerts a selective effect on the SNc area as our immunohistochemistry results indicated, we conducted molecular analysis in the midbrain rather than the specific SNc region due to technical limitations. More narrowly SNc-restricted analysis therefore might be necessary for future investigation. These researches, with the current study, will further consolidate the validity and utility of the repeated ECS treatments for various neuropsychiatric diseases including PD.
Although this study has focused on the examination of autophagy signaling alterations, the involvement other mechanisms could not be excluded. Accumulating evidence has suggested that neurotrophic factors regulating neuronal survival and brain development could play important role of the therapeutic action of the ECT. Animal studies have shown that repeated ECS treatments increase neurogenesis and activate proliferative signals such as ERK pathway and Cdk2-pRB-E2F1 cell cycle pathway and in the rat brain [64-66]. ECS-induced neurotrophic factors, such as brain-derived neurotrophic factor, can mediate anti-depressive and neuroprotective effects [63,66-68]. In addition to the biochemical changes, other studies hypothesized that neurophysiological changes, such as changes in cerebral blood flow, glucose metabolism, permeability of blood brain barrier, and neuroplastic changes such as alterations in volume of brain substructures may be responsible for the clinical effect of ECT [69]. Further studies will be necessary to investigate the involvement of those mechanisms in the therapeutic action of ECT on PD.
In this study, we clearly demonstrated that repeated ECS treatments normalized the Parkinsonian behavioral changes and prevented SNc DA cell loss in MPTP mice PD animal model. Moreover, ECS ameliorated the dysfunctions in the autophagy signaling in brain region-specific fashion. Repeated ECS treatments activated AMPK-autophagy signaling in the MPTP PD model mice, which could also promotes neuroprotective activity. In conclusion, the findings revealed the therapeutic effects of repeated ECS treatments on PD, which could be attributed to the neuroprotective effect of ECS mediated by AMPK-autophagy signaling.